
Remember me
In this appendix we justify the use of model (1) for the \(\Delta\)QTc Endpoint in parallel and crossover thorough QTc trials.
For a parallel group thorough QTc study, one can use the model
$$\begin X_= \pi _t + \vartheta _1 C_ + \vartheta _2 C_ +W_ + \varepsilon _ \end$$
(A1)
for the original QTcF observations \(X_\) for subject k in treatment arm \(\ell\) at time \(t = t_, \dots , t_\) pre-dose or \(t = t_1, \dots , t_N\) post-dose. The \(C_\) and \(C_\) are the corresponding PK concentrations for the two compounds, with \(C_=0\) and \(C_=0\) for all pre-dose time points, or for placebo (\(\ell =0\)). The fixed effects \(\pi _t\) describe the diurnal variation at these time points, and \(\vartheta _1\) and \(\vartheta _2\) are population slopes. The random subject effects \(W_\) are assumed to be independent normally distributed with mean 0 and variance \(\sigma _W^2\). The residual noise \(\varepsilon _\) terms are also assumed to be independent normally distributed with mean 0 and variance \(\sigma _\varepsilon ^2\). As usually, random effects and random noise are assumed to be independent as well.
The primary endpoint \(\Delta\)QTc is defined as
$$\begin Y_= X_-\sum _^X_ \end$$
(A2)
for all post-baseline time points \(t = t_1, \dots , t_N\). Using model (A1)and \(C_=0\) and \(C_=0\) for all pre-dose time points one gets
$$\begin Y_= \mu + \pi _t + \vartheta _1 C_ + \vartheta _2 C_ +U_ + \varepsilon _ \; , \end$$
(A3)
where \(\mu =-\sum _^\pi _\) and \(U_ =-\sum _^\varepsilon _\).
The parameter \(\mu\) in the model means that there will be a column in the design matrix with all entries equal to 1. This column is the sum of the columns in the design matrix that correspond to the parameters \(\pi _t\), which means that the model is over-specified. Hence, the parameter \(\mu\) is redundant and may be dropped from the model, resulting in model (1).
Most importantly, the original random subject effects \(W_\) cancel out. The new random effects \(U_\) have a much smaller variance (equal to \(\sigma _U^2=\frac\sigma _\varepsilon ^2\)) as the original random effects. This justifies our choice of \(\sigma _U^2\) in the simulation settings.
For a cross-over thorough QTc study, one can use the model
$$\begin X_= \alpha _i + \beta _j + \pi _t + \vartheta _1 C_ + \vartheta _2 C_ +W_ + \varepsilon _ \end$$
(A4)
for the original QTcF observations \(X_\) for subject k in sequence i and period j at time \(t = t_, \dots , t_\) pre-dose or \(t = t_1, \dots , t_N\) post-dose. The concentrations \(C_\) and \(C_\) are again equal to zero for all pre-baseline time points and placebo. The meaning of the other parameters is as explained after (A1). The random subject effect \(W_\) are again assumed to be independent normally distributed with mean 0 and variance \(\sigma _W^2\), and the residual noise \(\varepsilon _\) terms are assumed to be independent normally distributed with mean 0 and variance \(\sigma _\varepsilon ^2\). Note that pre-baseline values are obtained in each period here.
The primary endpoint \(\Delta\)QTc is defined as
$$\begin Y_= X_-\sum _^X_ \end$$
(A5)
for all post-baseline time points \(t = t_1, \dots , t_N\) in each period. Using model A4 and \(C_=0\) and \(C_=0\) for all pre-dose time points one gets
$$\begin Y_= \mu + \pi _t + \vartheta _1 C_ + \vartheta _2 C_ +U_ + \varepsilon _ \; , \end$$
(A6)
where \(\mu =-\sum _^\pi _\) and \(U_ =-\sum _^\varepsilon _\).
The parameter \(\mu\) is redundant and can be dropped from the model again. The cross-over specific fixed effects for sequence and period have canceled out, and the remaining model parameters are the same as for the parallel group design.
More importantly, the original random subject effects \(W_\) cancel out and are replaced by random subject by period interaction effects \(U_\). This implies that two observations that come from one subject but different periods are independent. One can therefore re-group the data into treatment groups of independent patients, and drop the sequence and the period index from the model. The data can be analyzed as if they had originated from a parallel group trial using model (1). Note that this requires that there are period-specific baseline values, and that the primary endpoint \(\Delta\)QTc is calculated using these period specific baseline values.
A.2: Justification of the model for the motivating exampleIn this appendix we derive the model (2) for the \(\Delta\)QTc endpoint in a dose-escalation trials. For a dose escalation study like that described in Section “Motivating example”, one can use the model
$$\begin X_= \pi _t + \vartheta _1 C_ + \vartheta _2 C_ +W_ + \varepsilon _ \end$$
(A7)
for the original QTcF observations \(X_\) for subject k from day d at time \(t = t_1, \dots , t_N\) post-dose. The \(C_\) and \(C_\) are the corresponding PK concentrations for the two compounds, with \(C_=0\) and \(C_=0\) for the pre-dose day (\(d=-1\)). The fixed effects \(\pi _t\) describe the diurnal variation at these time points, and \(\vartheta _1\) and \(\vartheta _2\) are population slopes. The random subject effects \(W_\) are assumed to be independent normally distributed with mean 0 and variance \(\sigma _W^2\). The residual noise \(\varepsilon _\) terms are also assumed to be independent normally distributed with mean 0 and variance \(\sigma _\varepsilon ^2\). As usually, random effects and random noise are assumed to be independent as well.
The primary time-matched \(\Delta\)QTc endpoint is defined as
$$\begin Y_= X_-\sum _^X_ \end$$
(A8)
for all post-dose time points \(t = t_1, \dots , t_N\) on all post-dose days. Using Eq. A7 and \(C_=0\) and \(C_=0\) one gets
$$\begin Y_= \vartheta _1 C_ + \vartheta _2 C_ +U_ + \varepsilon _ \; , \end$$
(A9)
where \(U_ =-\varepsilon _\).
Model (A7) assumes that the diurnal variation is the same on all dose escalation days. Hence, the diurnal variation has canceled out in model A9. Often, however, there are differences in study conduct between the pre-dose day \(d=-1\) on one end and the post-baseline days on the other. For example, there are usually no blood samples obtained on the pre-dose day. These differences may lead to differences in the diurnal variation between the pre- and the post-dose days. Allowing for such differences leads to model (2).
One could have used change from mean baseline as the primary endpoint in this study, with mean baseline being the mean across the pre-dose observations on day \(d=1\) here. The resulting model would have been similar, with a random subject effect \(U_k\) instead of a random subject-by-time interaction \(U_\). However, it is important to note that the pre-dose baseline values on day \(d=8\) should not be used to correct for baseline, as these observations already may be impacted by a potential QT effect induced by the drug.
A.3: Statistical model for pooled datasetsIn this appendix we briefly describe how to adapt the models for data pools. In the case of data pools, one can use the model
$$\begin X_= \pi _ + \vartheta _1 C_ + \vartheta _2 C_ + V_s + W_ + \varepsilon _ \end$$
(A10)
for the original QTcF observations \(X_\) for subject k from group \(\ell\) in study s at time at time \(t = t_, \dots , t_\) pre-dose or \(t = t_1, \dots , t_N\) post-dose (in the case of a parallel group trial). The diurnal variation \(\pi _\) is modeled as a study-by-time interaction term now, because the diurnal variation is a reflection of biology and study conduct. Therefore, even if two studies share the same time points, the diurnal variation may still differ. The term \(V_s\) describes a random study effect, assumed to be normally distributed with mean zero and variance \(\sigma _V^2\). The meaning of all other terms is obvious and as in the previous subsections.
Defining time-matched \(\Delta\)QTc as
$$\begin Y_= X_-\sum _^X_ \, , \end$$
(A11)
we get the model
$$\begin Y_= \pi _ + \vartheta _1 C_ + \vartheta _2 C_ +U_ + \varepsilon _ \end$$
(A12)
all post-dose time points \(t = t_1, \dots , t_N\). This is essentially the same calculation as for parallel group trials in Subsection A.1. Note that the random study effect \(V_s\) cancels out.
If one applies the approach to a crossover study that is to be added to the pool, the same model will evolve. Random study effects that are added on the level of the QTcF observations cancel out and do not appear in the derived model for the \(\Delta\)QTc observations. Therefore, model (A12) can be used for data pools, regardless of the design of the underlying studies.
A.4: Details on the simulation settingsTable 1 summarizes the basic settings for the simulation study described in Section “Results”.
Table 1 Estimates of diurnal variation, mean concentration profiles (ng/mL), and residual noise for parent and metabolite as used in the simulationsTable 2 shows the exact parameter values for four different scenarios where the supra-therapeutic dose has concentrations which are two-fold as compared to the therapeutic dose (\(\phi =2\)).
Table 2 Parameter values for \(\vartheta _1\) and \(\vartheta _2\) by sub-scenario for all four scenariosFig. 3
Contour lines for the subscenarios of Scenarios 1 and 3
In Fig. 3 one can see the \(10\,\)msec contour lines for the different sub-scenarios of scenario 1 and 3. These contour lines correspond to the pairs of concentrations \((c_1,c_2)\) of parent and metabolite where - given the parameters that drive the sub-scenario - the effect equals \(10\,\)msec. The parameters for sub-scenario 1 are chosen such that the contour line touches the alternative hypothesis space, which is the square with corner points (0, 0), \((0, C_)\), \((C_,0)\), and \((C_,C_)\). For sub-scenario 1, contour lines corresponding to a QT effect \(>10\,\)msec do not intersect with this square, whilst contour lines corresponding to QT effects \(<10\,\)msec intersect with this square. The \(10\,\)msec contour lines for the other scenarios do not intersect with the alternative hypothesis, and they are more and more distant when scenario numbers increase. You can see that for scenario 1, where both \(\vartheta _1\) and \(\vartheta _1\) are positive, the maximum effect on the alternative is reached in the corner point \((C_,C_)\), whereas for scenario 3, where \(\vartheta _1\) is negative, the maximum on the alternative hypothesis is achieved at the corner point \((0, C_)\).
A.5: Details on the motivating exampleIn this appendix we provide additional details on the study described in Section “Motivating example”.
Figure 4 shows the PK profiles of parent drug and the second metabolite alongside the QTc profile for the drug. It can be seen that the maximum QTc effect is achieved at a later time point as compared to when the maximum concentration of the parent drug is achieved. This may erroneously be interpreted as hysteresis when considering only the parent drug. In a situation with two active compounds, mis-alignment of the times of maximum QT effect and maximum PK effect may just reflect that the two active compounds achieve there maximum concentration at different time points.
Fig. 4
Concentration of parent drug (yellow) and metabolite (blue) (top, log scale) and time-matched \(\Delta\)QTcF (bottom) on day 8
In the case of our study there were actually three potentially active metabolites. In principle, all of them should be included in the exposure-response model. However, if the profiles are two similar (like parent and first metabolite or second and third metabolite, see Fig. 5) one cannot really separate the effects of these colinear compounds. One approach to deal with such a situation is to drop some of the colinear compounds from the model (see, for example, [15]). We planned to use parent and second metabolite for the primary analysis, and to repeat the same analysis for other pairs of compounds (parent with third metabolite, first and second metabolite, etc.) as sensitivity analyses.
Fig. 5
Concentration of parent drug and all three metabolites on day 8
Similarly, we planned to pool the data from day 1 and the data obtained under steady state (day 8) for the primary analysis, and to use model (2) as the primary model. Identical analyses just with the data from days 1 or 8 or using model (2) with an additional interaction term \(\vartheta _C_C_\) were planned as sensitivity analyses again.
The estimated slope for the parent drug was \(\hat_1=-0.00047\), indicating that the parent drug has no effect or very little effect on the QTc interval. In contrast, the corresponding slope estimate for the metabolite is \(\hat_2=0.000095\). The maximum concentration for this compound occurs around 4 hours post dose (on day 8) when there is still a considerable amount of parent drug in the blood. Therefore, the negative slope estimate of the parent drug and the increasing effect of the metabolite counterbalance each other. As a consequence, the maximum QTc effect occurs after only after 10 hours when the concentration of the metabolite is still high, but the concentration of the parent compound is already very low again (see Fig. 4).
Overall, the impact of the metabolite on the QT interval appeared to be mild. This can be seen in Fig. 6, where the pairs of PK concentrations are shown together with the \(10\,\)msec contour line \(10=\hat_1 C_1 + \hat_2 C_2\) when using model (2). All observed data points are in the area where the effect is below \(10\,\)msec, and actually well below this threshold as all data points are distant from the \(10\,\)msec contour line. This was confirmed by the bootstrap analysis. The conclusions did not change when using Eq. 2 with an additional interaction term \(\vartheta _C_C_\), or when using other pairs of compounds.
Fig. 6
Summary of results: individual data points and \(10\,\)msec contour line when using model (2)
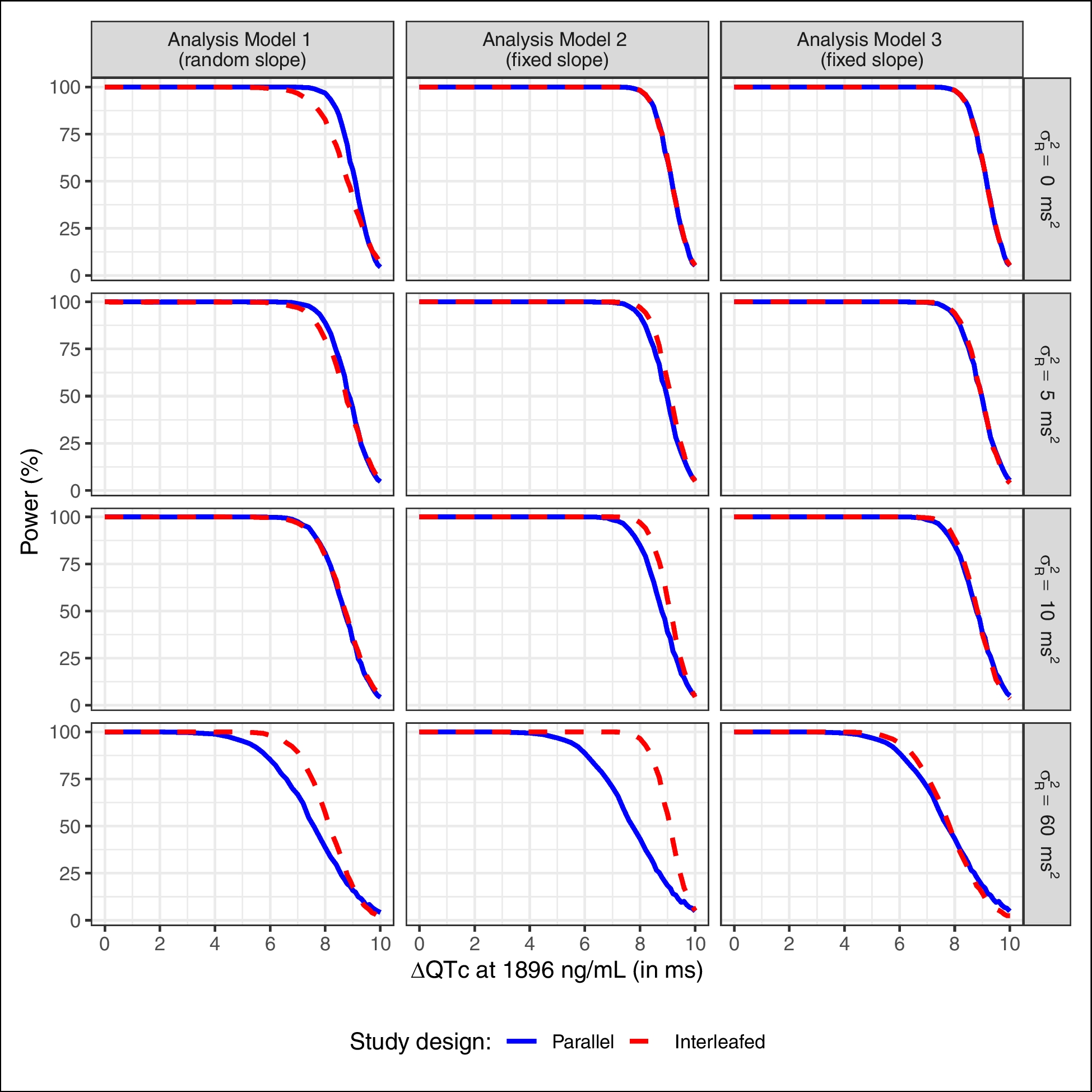
A.6: Additional simulation resultsThe additional figures presented in this appendix have been discussed in Section “Results”.
Fig. 7
Simulation results for scenarios 1 to 4 (\(\phi =2\)) for N=20 (dotted red line), 50 (dashed green line), and 100 (solid blue line); parent and metabolite correlated (\(\rho _=0.5\)), \(\sigma ^2_U=\frac\sigma ^2_\varepsilon\), and \(\sigma ^2_\varepsilon =50\)
Fig. 8
Simulation results for scenarios 1 to 4 (\(\phi =2\)) for N=20 (dotted red line), 50 (dashed green line), and 100 (solid blue line) with interaction term in the analysis; parent and metabolite uncorrelated (\(\rho _=0\)), \(\sigma ^2_U=\frac\sigma ^2_\varepsilon\), and \(\sigma ^2_\varepsilon =50\)
Fig. 9
Simulation results for scenario 1 (\(\phi =2\)) for N=20 (dotted red line), 50 (dashed green line), and 100 (solid blue line); parent and metabolite uncorrelated (\(\rho _=0\), top row) and correlated (\(\rho _=0.5\), bottom row); \(\sigma ^2_U=\sigma ^2_\varepsilon\) (left column) and \(\sigma ^2_U=3\sigma ^2_\varepsilon\) (right column); \(\sigma ^2_\varepsilon =50\)
Fig. 10
Simulation results for scenario 1 (\(\phi =2\)) for N=20 (dotted red line), 50 (dashed green line), and 100 (solid blue line); parent and metabolite uncorrelated (\(\rho _=0\), top row) and correlated (\(\rho _=0.5\), bottom row); \(\sigma ^2_U=\frac\sigma ^2_\varepsilon\); \(\sigma ^2_\varepsilon =75\) (left) to \(\sigma ^2_\varepsilon =100\) (right)
Comments (0)