
Remember me
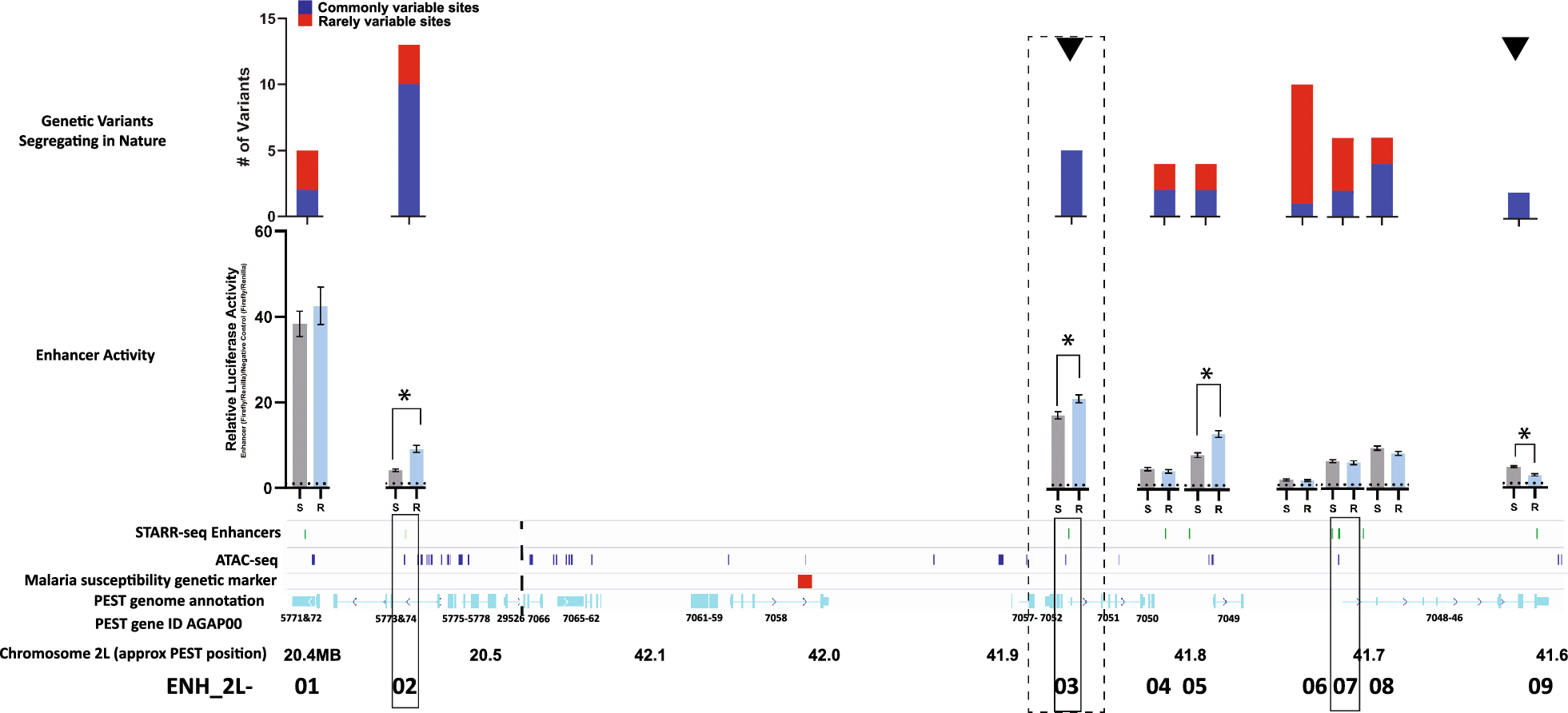
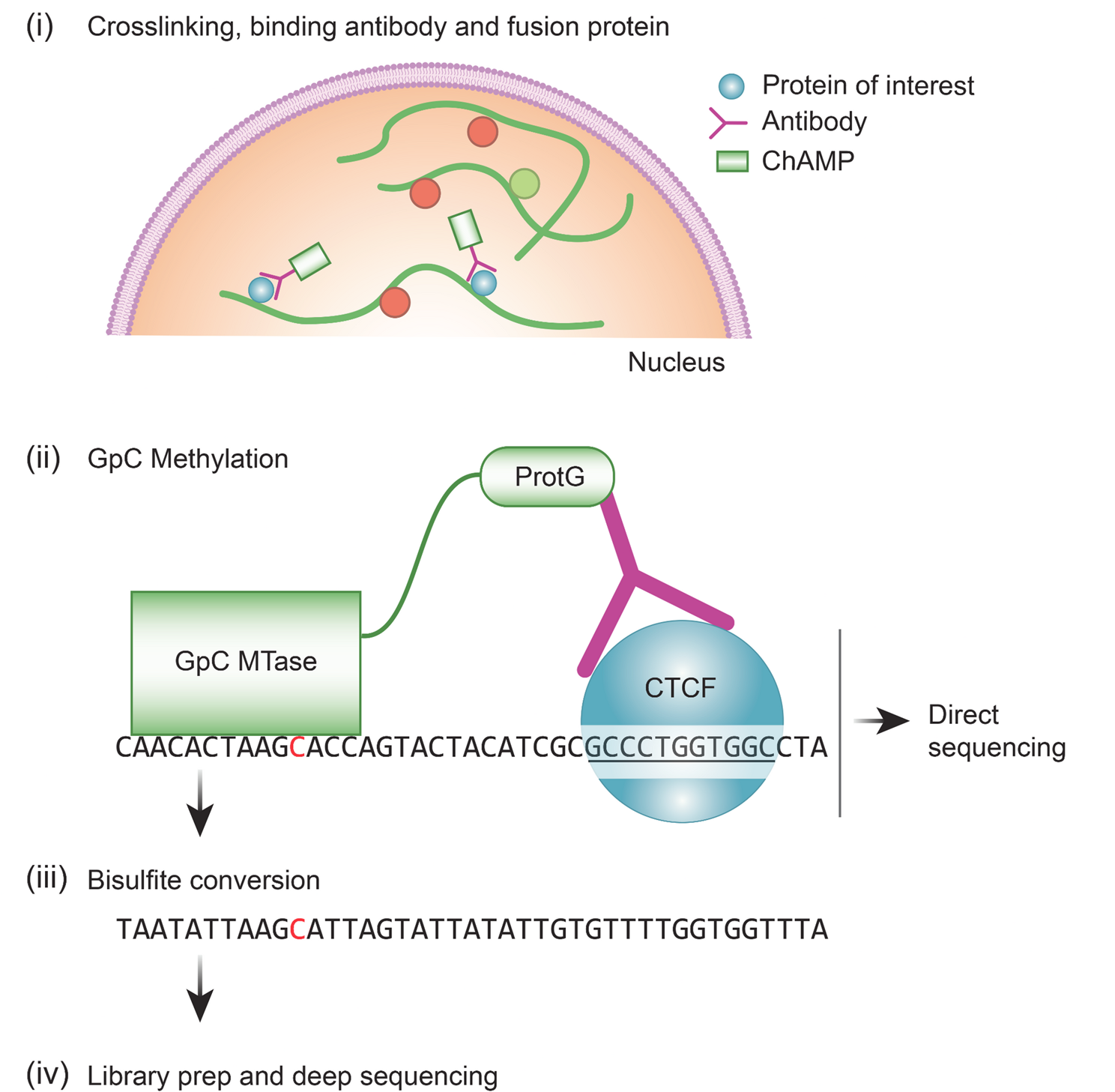


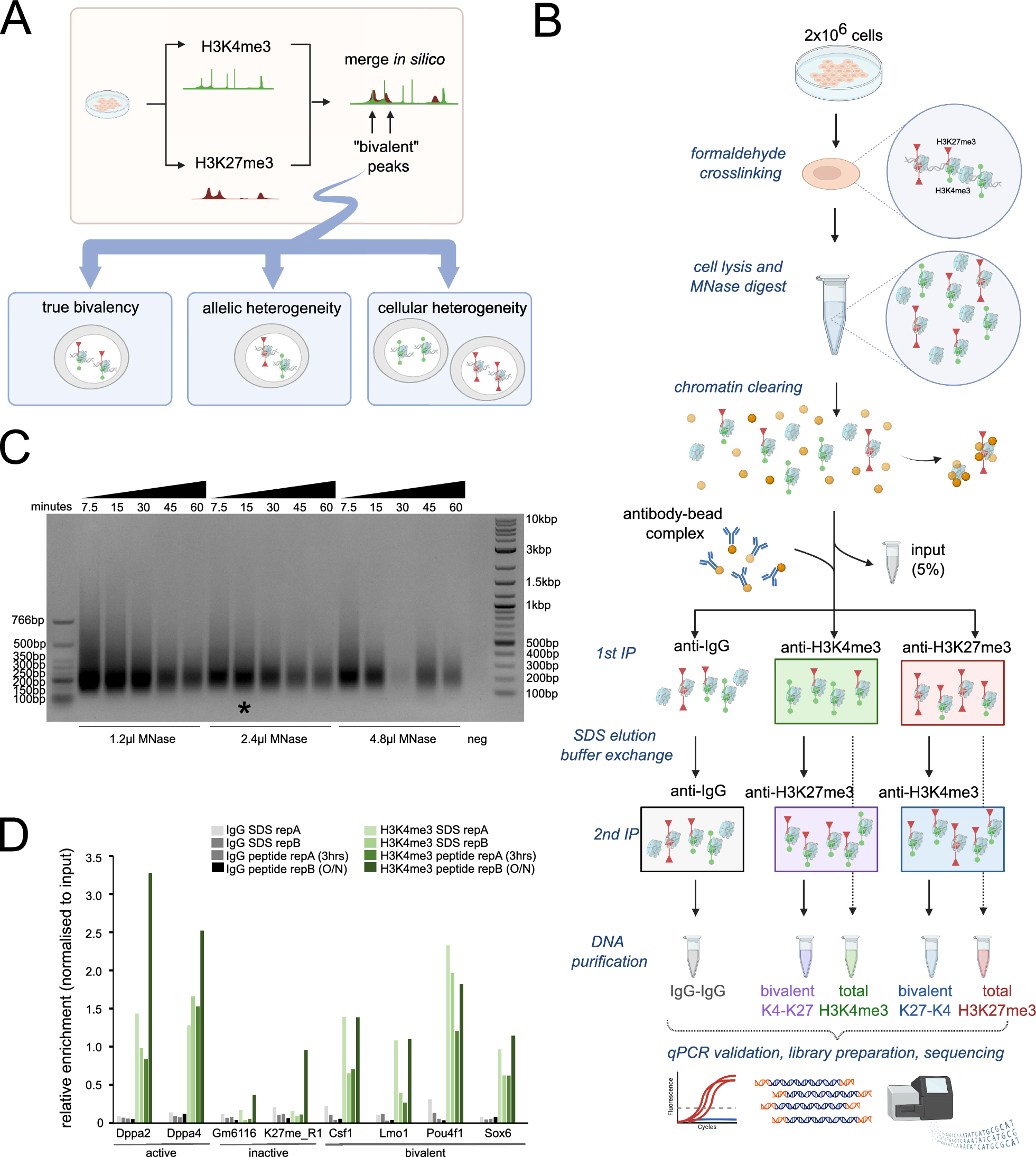
We envisioned that a protein capable of binding primary antibodies, switching-on its enzymatic activity and distinctly modifying adjacent DNA, can provide an alternative method to ChIP-seq in the characterization of protein binding sites (Graphical abstract and Fig. 1A). The advantages of this approach include (i) avoidance of the sample loss inherent in the immunoprecipitation methods (ii) multiple and bi-modal adjacent DNA modifications from a single enzyme-molecule can allow for a confident detection from a single sequenced molecule (iii) the primary antibody only needs to bind to its target, and not immunoprecipitate it, enabling the use of antibodies with weaker binding capacities (iv) modifications can be directly identified from single long molecules, enabling the exploration of 3D folding and the relations between adjacent sites (v) development of new MTases with a different recognition sequence can allow the multiplexing of several DNA-binding proteins on a single molecule. This is similar to DiMeLo-seq, however, the use of GpC MTase keeps this method compatible with bisulfite sequencing.
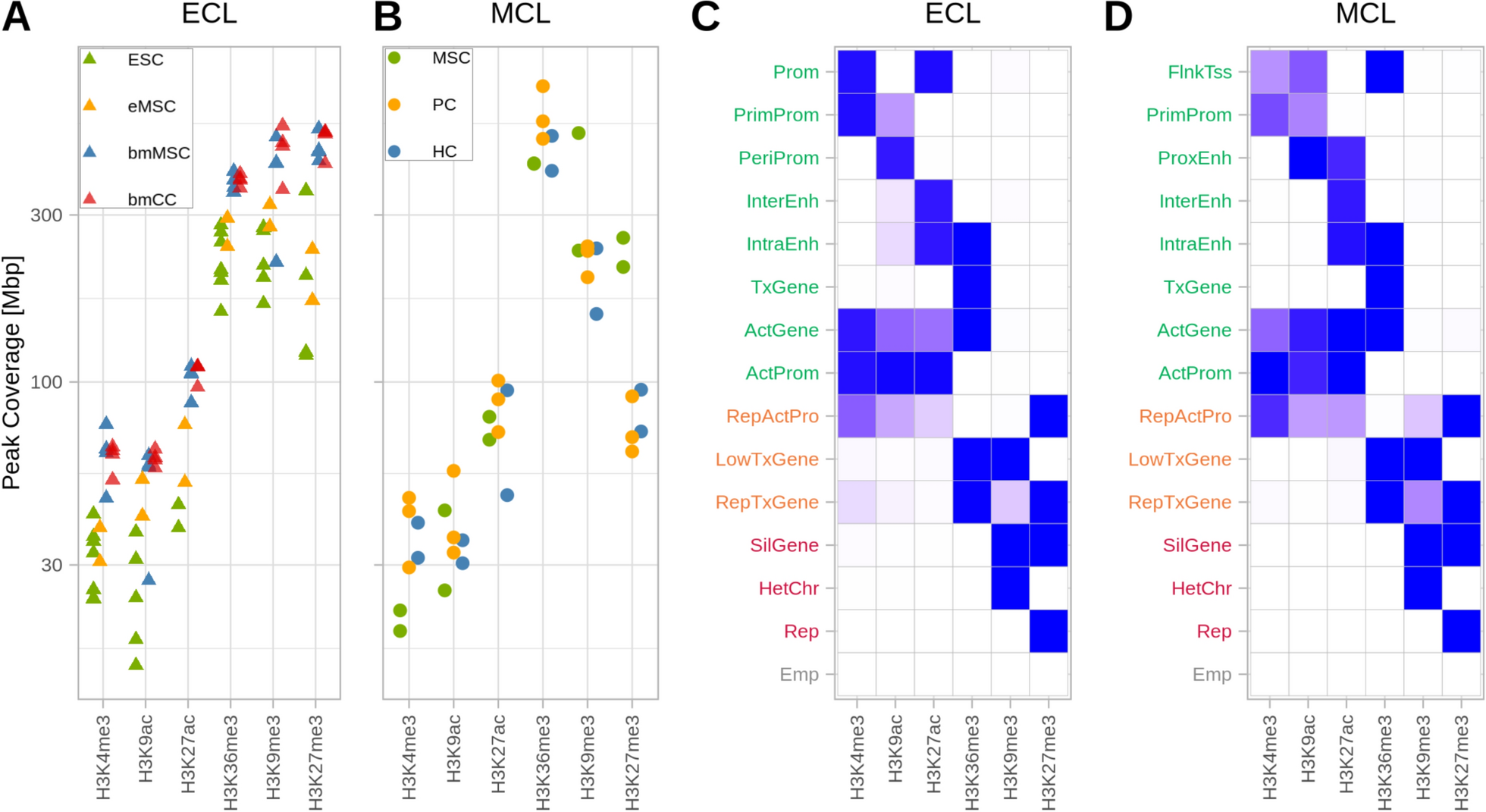
Method developmentWe designed (Fig. 1A and supplementary data), expressed (Fig. 1B) and purified (Fig. 1C) a fusion protein that (a) binds to antibodies and (b) methylates GpC in the presence of S-Adenosyl methionine (SAM). We validated that the fusion protein binds to antibodies in-vitro and in situ (Fig. 1B and D) and is able to GpC methylate purified DNA in-vitro (Fig. 1E, F).
Next, we developed (Supp. Figure 1A-C; Supp. Figure 2; Supplementary protocol) and optimized a protocol for antibody-guided methylation of fixed samples. As a proof-of-concept, we focused on the well characterized CCCTC-binding factor (CTCF) in HeLa cells. Standard formaldehyde fixation significantly inhibited DNA methylation in situ. However, short fixation time with low formaldehyde concentration enabled enzyme mediated methylation (Supp. Figure 1B). Methylation was further enhanced by a brief heating of the fixed samples prior to adding the enzyme (Supp. Figure 1C).
Fig. 1
ChAMP specifically methylates GpC near the proteins of interest. (A) diagram of ChAMP design. (B) Western blot of lysed T7 Express lysY E. coli expressing ChAMP show direct binding of the secondary antibody. No primary antibody was used (see Material & Methods) (C) Protein staining of HIS-tag purified ChAMP. (D) Detecting in-situ antibody binding. Immunofluorescence of HeLa cells stained with DAPI, a primary rabbit anti lamin B antibody and (i) positive control - AF555 anti-rabbit (first panel) or (ii) negative control - Cy3 anti mouse (no binding expected due to mismatch between primary and secondary species - second panel; inset at higher exposure shows no weak lamina staining) or (iii) ChAMP plus Cy3 anti mouse (bridged binding - third panel). As ChAMP has multiple IgG binding domains, it can bridge the rabbit and anti-mouse antibodies in panel 3, visualizing the nuclear envelope and proving correct localization. Scale bar − 10 μm (E) Detecting GpC methylation. GpC methylation inhibits HaeIII digestion. A PCR product containing multiple HaeIII cut sites was treated with either a lysate or purified ChAMP, in the presence of SAM, and digested with HaeIII. 3 purification methods were tested (see Material & Methods). Digested product was resolved on a gel. (F) Validation of GpC methylation in-vitro by ChAMP. Text shows genomic sequence, arrows point to GpC. Top diagram - Purified DNA was incubated with the ChAMP, bisulfite converted and Sanger sequenced. Bottom diagram - Validation of GpC methylation in-situ by ChAMP. Fixed cells were target methylated with ChAMP, DNA extracted, bisulfite converted and amplified by PCR. Significant methylation only seen upon mild fixation conditions. (G) Left - Restriction-PCR design overview. Genome track showing the targeted CTCF peak, HaeIII cut sites (estr enzymes - black lines) and PCR primers (red arrows). Right - ChAMP can specifically methylate DNA. Fixed cells treated with the ChAMP protocol showed a strong band following restriction-PCR, however no band at the correct size was seen when ChAMP was omitted and only a weak band when the CTCF antibody was omitted. Following the methylation reaction, DNA was digested with HaeIII and PCR reaction with limited number of cycles amplified the DNA products. Schematic representation of the method, applied to CTCF
Fig. 2
(A) Frequency of GpC methylation peaks detected by ChAMP, aligned to the center of CTCF ChIP-seq peaks (position 0; filtered by minimum of 7 events in 200 bp of sliding window moving average). The results show strong enrichment of methylation near known CTCF binding sites, indicating successful detection of CTCF-DNA interactions. In contrast, control conditions in which either ChAMP or the CTCF antibody was omitted show no significant enrichment, confirming the specificity of the method. (B) Zoom-in on ChAMP without the density filter showing a double peak. (C) Post-amplification enrichment - Schematic overview of the post-enrichment protocol. After bisulfite conversion, DNA is amplified by PCR for 1–5 rounds with random primers that do not introduce GpC. The purified product is further amplified, introducing adapters that lack GpC. The resulting product is re-methylated with the ChAMP protein. As this is amplified bisulfite converted DNA, any GpC found represents a methylated GpC (including GpCpG) in the original sample. (D) Distance of GpCpH methylation relative to the center of the closest CTCF ChIP-seq peak using post-amplification enrichment
We then validated our ability to bind to primary antibodies of interest (Fig. 1D), to methylated CpG in-vitro (Fig. 1E, F), and to in situ methylate near a CTCF binding site using restriction-PCR and Sanger bisulfite sequencing (Fig. 1G). While methylation was enriched near the target of interest, nonspecific binding created background methylation (Supp. Figure 3A, lower left panel). Washes with detergents minimized nonspecific binding without affecting enzymatic activity (Supp. Figure 3A, B).
Methylation patternsNatural GpCpH (H being any nucleotide but G) methylation rarely occurs in mammalian cells, and is thereby easily distinguishable from existing methylation patterns. To assess data quality, we sequenced samples to low-coverage and mapped the GCH methylation distribution relative to the closest known CTCF binding site, selecting for conditions that minimize off-target methylation. Incomplete conversion of cytosine into uracil resulted in background (typically 0.5-1%), however, as unconverted cytosines distributed randomly, requiring multiple adjacent methylation events (henceforth dense methylation) removed much of the background. To map the methylation patterns from CHAMP, we sequenced the HeLa CTCF samples to a higher coverage (122 M mapped reads). We aligned all the called methylation events relative to known CTCF peak centers, then applied a sliding window threshold, requiring 7 events within 200 bases, resulting in a strong peak overlapping the center of CTCF peaks. This pattern was not observed when either ChAMP or the primary antibody were omitted (Fig. 2A). Zooming in without the sliding window, revealed a double peak ~ 30 bp on either side of the CTCF ChIP-seq peak center (Fig. 2B), presumably because CTCF and the bound antibody-ChAMP complex limit enzyme accessibility to adjacent DNA, as observed by in similar conditions [12]. Weak secondary peaks, approximately 220 bp from peak center, may be the result of nucleosome occupancy (see below).
To evaluate ChAMP’s ability to identify protein binding sites, we developed a basic peak calling strategy for initial comparisons with established methods. Methylation events were expanded using bedtools slop and called using callpeaks2.pl (https://github.com/Henikoff/Cut-and-Run/). However, more sophisticated algorithms that account for the unique properties of antibody-guided methylation will be needed for optimal peak identification, as discussed below.
Methylation enrichmentWithout target enrichment, ChAMP requires genome-wide coverage. While this provides the additional information of bound/unbound ratios, it requires deeper sequencing than standard ChIP-seq experiments. Enrichment for methylated DNA can reduce the sequencing requirements. However, target enrichment may involve primary substance losses, thus inhibiting our ability to apply it to small samples. We circumvented this limitation by creating a post-amplification library enrichment protocol that enriches for GpC methylation, but not CpG (Fig. 2C). In our bisulfite converted library-preparation protocol we have three consecutive PCRs, used for random priming, amplification and indexing, respectively [9]. We modified the primers used in the first two PCR reactions to preclude the introduction of GpCs. Following the bisulfite conversion and amplification PCR, the only remaining GpCs present in the sample originated from a methylated GpC. We then re-methylated the sample (see Supplementary protocol) and immunoprecipitated the methylated DNA, effectively enriching for molecules that were originally labeled. We then performed a final amplification with indexing PCR prior to sequencing. Following the enrichment protocol, a clear structure matching nucleosome-free DNA surrounding the CTCF binding sites was seen in the cumulative data (Fig. 2D). Mapping methylation sites revealed that these predominantly overlap the CTCF ChIP-seq and Cut&Run signal (Fig. 3A, B). To validate the generality of ChAMP we tested it with an antibody directed to the histone modification H3K27ac. Dense methylated sites yielded methylation patterns that overlapped with a ChIP-seq signal (Fig. 3C). Additional methylation sites often overlapped DNase hypersensitive regions or were adjacent to exons, suggesting the possibility of 3D folding.
Fig. 3
(A) Genome browser track showing dense methylation events for enrichment ChAMP using a CTCF antibody. These methylations partially co-localize with CTCF Cut&Run and ChIP-seq. (B) Zoom-in on a ChIP-seq peak. (C) Genome browser track showing dense methylation events for ChAMP using a H3K27ac antibody. These methylations partially co-localize with H3K27ac ChIP-seq
Fig. 4
(A) Genome browser track showing methylation probability of k-mers along a single ~ 150 kb molecule from a ChAMP experiment using an H3K27ac antibody. Methylation probability was enriched, but not exclusive, to ChIP-seq peaks, open chromatin and ChromeHMM Active Promoters. (B) Zoom-in on a ChIP-seq peak. HeLaS3 H3K27ac - ChIP-seq signal and peaks; HeLaS3 ChromHMM - chromatin state track; HeLaS3 DS - DNaseI HS Density Signal
Single molecule mapping of binding sitesVarious single molecule sequencing technologies allow direct detection of DNA modifications [14, 15]. We used the modified version of nanopolish that is trained to call GpC methylation from Lee et al. [10, 12]. Nanopolish uses a hidden Markov model trained on enzymatically methylated DNA to distinguish between cytosine and 5-methylcytosine. While single molecule sequencing and methylation detection have higher error rates than clonal amplification and sequence by synthesis, it increases the maximal read length by 3 orders of magnitude, to over 100 kb. At these scales, the extended methylation pattern resulting from a single bound site can be seen on a single molecule. Moreover, the relationship between the occupancy of neighboring sites and how distance affects it can be analyzed. Despite using fixed cells, we were able to obtain multi-kilobase sequences with good quality scores (Supp. Figure 4; N50: 8327 bp; [16]). For each read, we calculate the GpC methylation probabilities for k-mers along the sequence [12]. Methylation was greatly enriched near known H3K27ac binding sites, with multiple adjacent k-mers showing high probability of methylation (Fig. 4A, B).
Comments (0)