The pathology of synonymous variants can be complex, with multiple possible causes of pathology such as alterations in splicing, protein stability, and translation (Hunt et al. 2014; Lin et al. 2023). However, in the case of GSDME, the gain-of-function mechanism reflects a simpler pathology of exon skipping that generates a transcript that lacks exon 8. This lack of exon 8 results in the formation of a constitutively active protein that results in apoptosis. This pathophysiology has been verified in both cell culture (de Beeck et al. 2012) and mouse models (Xiao et al. 2024). This common pathophysiology allows one to assume that all pathogenic SNVs involve aberrant splicing and thereby eliminates the need to investigate other factors such as changes in mRNA structure and stability, miRNA binding, and codon usage.
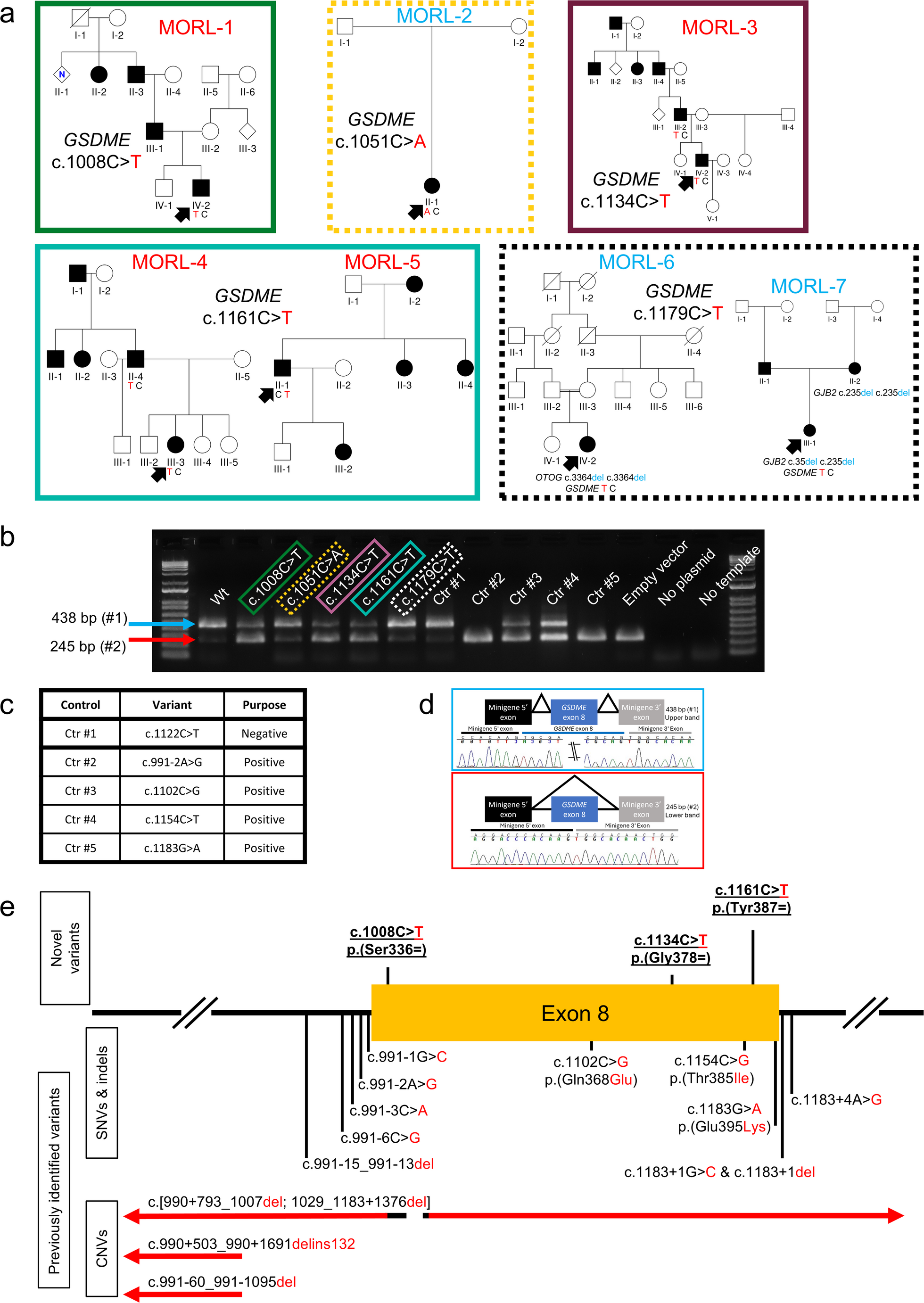
These results are consistent with our hypothesis that there are pathogenic synonymous variants that were otherwise being filtered out. Previously, all known pathogenic GSDME variants were in the intronic regions flanking exon 8, missense variants within exon 8, or structural variants. Our identification of the first pathogenic synonymous GSDME variants illustrates the importance of assessing synonymous variants in the context of genetic diagnostics. Consistent with previous work (Booth et al. 2018), we also observe splice-altering variants outside the proximity of the exon boundaries (Fig. 1e, Supplementary Fig. S4). These results highlight the need to assess variants for alterations in splicing regardless of their location within the exon and the need for in silico prediction tools that are not constrained to the proximity of splice acceptor and donor sites.
The recurrence of the c.1161 C > T, p.(Tyr387Tyr) variant in two unrelated probands is not entirely unexpected (Fig. 1a). Despite the relative rarity of GSDME as a cause of HL (< 1% of genetically diagnosed cases) (Sloan-Heggen et al. 2016), there are other recurring pathogenic variants such as the c.991–2 A > G (Booth et al. 2018) and the c.991 − 15_991-13del (Booth et al. 2020) variants. Furthermore, the polypyrimidine tract of GSDME intron 7 is a mutational hotspot (Booth et al. 2020), which raises the possibility of the existence of other mutational hotspots in the same gene. This presence of rare, but recurring variants indicates the need for functional assessment of variants regardless of their rarity.
Our observations of varying levels of aberrant splicing in minigene is not unexpected given that it has already been reported that the c.991–6 C > G variant results in less of the truncated transcript compared to the c.990 + 503_990 + 1691delins132 variant (Bischoff et al. 2004). Many genes implicated in HL have genotype-phenotype correlations and/or population-level differences (Walls et al. 2020). It is logical to hypothesize that greater levels of aberrant splicing will corelate with greater levels of mutant protein, and by extension more hair cell death and more severe HL. Audiometric analysis supports this hypothesis. Complete loss of splicing results in more severe HL compared to partial loss of splicing, as reflected in more severe HL at a given age and/or more rapid progression at the middle and high frequencies (Fig. 2). The lack of statistically significant differences in the severity of HL at the low frequencies may be due to the lack of statistical power. The HL is mild at the low frequencies (Thorpe et al. 2022) and the differences at 250 and 500 Hz are very small (Fig. 2b-c).
While some HL genes such as TECTA and WFS1 have ethnic-based differences (Walls et al. 2020), they are not observed for GSDME. The c.991–6 C > G and c.990 + 503_990 + 1691delins132 variants were both observed in large Dutch families (Bischoff et al. 2004) and demonstrate results consistent with complete loss of splicing causing more severe HL (Supplementary Fig. S3). These observations also are consistent with the report that persons carrying the c.991–6 C > G variant have better speech recognition than persons carrying the c.990 + 503_990 + 1691delins132 variant (Bischoff et al. 2004) and are consistent with a prior investigation of the GSDME c.991 − 15_991-13del variant that was unable to find ethnic-based HL variability (Booth et al. 2020). Taken together, these findings suggest that the primary driver of differences in the severity of GSDME-related HL is a variant-specific effect.
We also observe that of the three GSDME variants analyzed for genotype-phenotype correlations, the c.991 − 15_991-13del variant has the mildest HL (Supplementary Fig. S3). Presumably, this would indicate that the c.991 − 15_991-13del variant has the least amount of aberrant splicing. Future work that evaluates the amount of aberrant splicing in a uniform manner could confirm this hypothesis. A detailed comparison of the precise levels of aberrant splicing would be of future interest because it could lead to the determination of the minimum amount of aberrant splicing required to cause HL and thereby guide potential therapeutic interventions such as knockdown of the mutant transcript.
In the context of GSDME-related HL, in silico prediction tools should be used cautiously. HSF, SpliceAI and SPiP in the context of the retrospective MORL and gnomAD cohorts are highly inaccurate (Figs. 4 and 5). This conclusion is consistent with other reports recommending in silico assessment to be used as a screening tool rather than as a diagnostic tool (Soukarieh et al. 2016; Moles-Fernández et al. 2018; Katneni et al. 2019; Walker et al. 2023; Sullivan et al. 2025). We do point out that HSF might be useful as a screening tool for splice-altering variants because it has a sensitivity of over 75% in the context of GSDME. In contrast, the utility of SpliceAI appears to be much more limited (Fig. 5).
We do note the reported accuracy in our cohorts is lower than that reported in other studies. This difference in accuracy may reflect the specific nature of GSDME, where the pathology excludes certain types of aberrant splicing such as the formation of cryptic splice acceptor and donor sites that are seen in other genetic disorders. The possibility of a gene-specific difference is not surprising, given that there are other deafness gene-specific differences such as in minor allele frequency thresholds (Azaiez et al. 2018). In addition, we investigated variants regardless of their location within an exon. In contrast, most studies and prediction tools prioritize investigating variants near the canonical splice donor and acceptor sites (Dionnet et al. 2020; Walker et al. 2023; Sullivan et al. 2025). Our observations of splice-altering variants outside the splicing consensus region (Supplementary Fig. S4) also prevented us from using commonly available and user friendly splice prediction tools such as MaxEntScan (Yeo and Burge 2004), dbscSNV (Jian et al. 2014), and SpliceAPP (Huang et al. 2024) because they lacked the ability to assess the majority of variants in our cohorts. These results indicate the need for in silico prediction tools that can assess variants for splice-altering effects regardless of their location in the genome.
The possibility of a bias due to a focus on variants near the canonical splice acceptor and donor sites would be consistent with HSF, SpliceAI, and SPiP appearing to be more accurate when predicting the impact of previously reported pathogenic variants (Supplementary Table 5). This bias presumably could be explained by the fact that both HSF, SpliceAI and SPiP are trained primarily on the normal splicing patterns observed with wild-type exons (Desmet et al. 2009; Jaganathan et al. 2019; Leman et al. 2022). However, one must be cautious when drawing conclusions from a retrospective assessment because of publication bias and data circularity. For example, the three missense variants were already flagged and prioritized for investigation by HSF at the time of their publication (Booth et al. 2018). It is also important to note that both HSF and SpliceAI have a false negative prediction for the c.991–6 C > G variant (Supplementary Table 5), despite the presence of extensive genetic evidence as well as harvested RNA to show that it is a pathogenic, splice-altering variant (Bischoff et al. 2004). We also highlight that in the context of the MORL cohort, a reported family history consistent with autosomal dominant HL had a stronger correlation with altered splicing than the HSF and SpliceAI predictions (Figs. 1a-b and 4b). These results indicate that in silico assessment cannot be used to override genetic evidence.
Previous work suggested using conservation as a way of prioritizing variants for their impact on splicing in the context of GSDME missense variants (Booth et al. 2018). Our results indicate that in the context of synonymous and intronic variants, there is no observable correlation between conservation and a variant’s impact on splicing (Fig. 4b and Supplementary Table 3). As a result, one should be cautious when using conservation as a method of screening for splice-altering variants.
In conclusion, the assessment of synonymous variants is an important part of genetic diagnostics. We expand the mutational landscape of GSDME-related HL to include synonymous variants. Given that there were 3 missense variants previously identified in the MORL cohort (Booth et al. 2018) and 3 synonymous variants identified in this study of the MORL cohort, we show that there is a near even split between pathogenic missense and synonymous GSDME variants. We also demonstrate that there is a recurring pathogenic synonymous variant. Finally, we identify that there are variant-dependent differences in aberrant splicing levels. These differences in aberrant splicing are reflected in the phenotype-genotype correlation of GSDME-related HL, where greater amounts of expressed mutant protein result in more severe and rapidly progressing HL in the middle to high frequencies. These differences in HL suggest that a knockdown strategy targeting the mutant transcript can reduce the impact of GSDME-related HL.
Identification of pathogenic variants remains a challenge, particularly when one is interested in identifying splice-altering variants that reside outside of the proximity of the canonical splice sites. One must consider the potential impact on splicing regardless of the variant’s in silico prediction, location within the exon, or conservation.


Comments (0)