
Remember me




Following the initial invasion, the merozoite becomes enclosed within a parasitophorous vacuole. During this early ring stage, biomechanical changes are minimal, and the RBCs retain much of their original deformability. However, as the parasite matures into the trophozoite stage, it exports numerous proteins to the RBC membrane and cytoskeleton. Among these proteins, knob-associated histidine-rich protein (KAHRP) and others promote the formation of knobs—surface protrusions that serve as platforms for endothelial adhesion. This process increases membrane rigidity by reorganizing cytoskeletal interactions, particularly among spectrin, actin, and anchoring proteins such as ankyrin [13, 22]. During this stage, iRBCs undergo extensive structural remodeling, reaching peak stiffness. The formation of knobs serves as anchoring points for P. falciparum RBC membrane protein 1 (PfEMP1), a major virulence factor that mediates adhesion to endothelial receptors [23]. The resulting increased membrane rigidity significantly impairs the ability of iRBCs to traverse narrow capillaries, leading to their sequestration within microvascular beds and helping them evade splenic clearance [24].
Cytoskeletal remodeling and its function in Plasmodium invasionThe parasite invasion process is highly dynamic and requires the RBC membrane to deform and wrap around the invading parasite. However, excessive membrane rigidity or elevated cortical tension in RBCs can significantly impair invasion. Biophysical studies have revealed a critical membrane tension threshold above which the parasite fails to enter. Recent theoretical and computational models have quantified this effect [6]. demonstrated that increased effective membrane tension and cortical shear resistance in red blood cells act as mechanical barriers to merozoite invasion. According to their findings, an increase in membrane tension can obscure the ability of merozoites to generate sufficient wrapping forces to successfully invade RBCs, thus blocking internalization.
An analytical model was developed by [7] that describes the mechanics of RBC membrane wrapping during active invasion. Their results indicate that parasite-driven forces alone are insufficient to complete membrane envelopment under high-tension conditions, suggesting that intrinsic RBC deformability is a determinant of invasion success. Together, these findings support the idea that RBC biomechanical properties are not passive substrates but active determinants of P. falciparum pathogenesis. Even with functional invasion machinery, the merozoite cannot overcome the physical resistance of overly stiff or tense red blood cells, underscoring the functional link between host cell deformability and parasite entry.
In this respect, a recent interesting study also demonstrated that specific alleles present in the RBCs of healthy donors are related to the prediction of parasite replication. Polymorphisms in proteins that make the backbone of the membrane flexible, ion channels related to hydration changes, and membrane proteins are related to parasite invasion. Although these polymorphisms do not trigger a pathology or disease in the donors, they do influence the ability to harbor the parasite that causes malaria [25]. Despite these data that have emerged in recent years, no clear evidence has been reported from any study that has tested whether this extensive phenotypic and genetic diversity in RBCs influences malaria susceptibility.
A growing body of evidence highlights how naturally occurring mutations in genes encoding RBC structural or regulatory proteins can modulate susceptibility to Plasmodium falciparum infection. In a recent comprehensive review [9], summarized multiple host variants that alter red blood cell biomechanical properties, thereby impairing parasite entry. Among these, the Dantu blood group stands out as a protective polymorphism caused by a structural rearrangement at the GYPA–GYPB locus. This event produces a chimeric glycophorin protein that increases membrane tension and significantly reduces invasion efficiency [26,27,28]. Another example is Southeast Asian Ovalocytosis (SAO), resulting from a 27-bp deletion in SLC4A1, which encodes Band 3. This mutation leads to RBCs with reduced deformability and oval morphology, conferring resistance to multiple P. falciparum strains; a common polymorphism in the mechanosensitive cation channel gene PIEZO1 (E756del) has been associated with reduced red cell volume and altered membrane properties and has been linked to protection from severe malaria in African populations [29, 30].
Functional studies using genetically modified mice provide additional evidence for the importance of membrane-linked structural proteins; for example, mutations in ankyrin impair red cell deformability and confer partial resistance to Plasmodium chabaudi invasion [31,32,33,34].
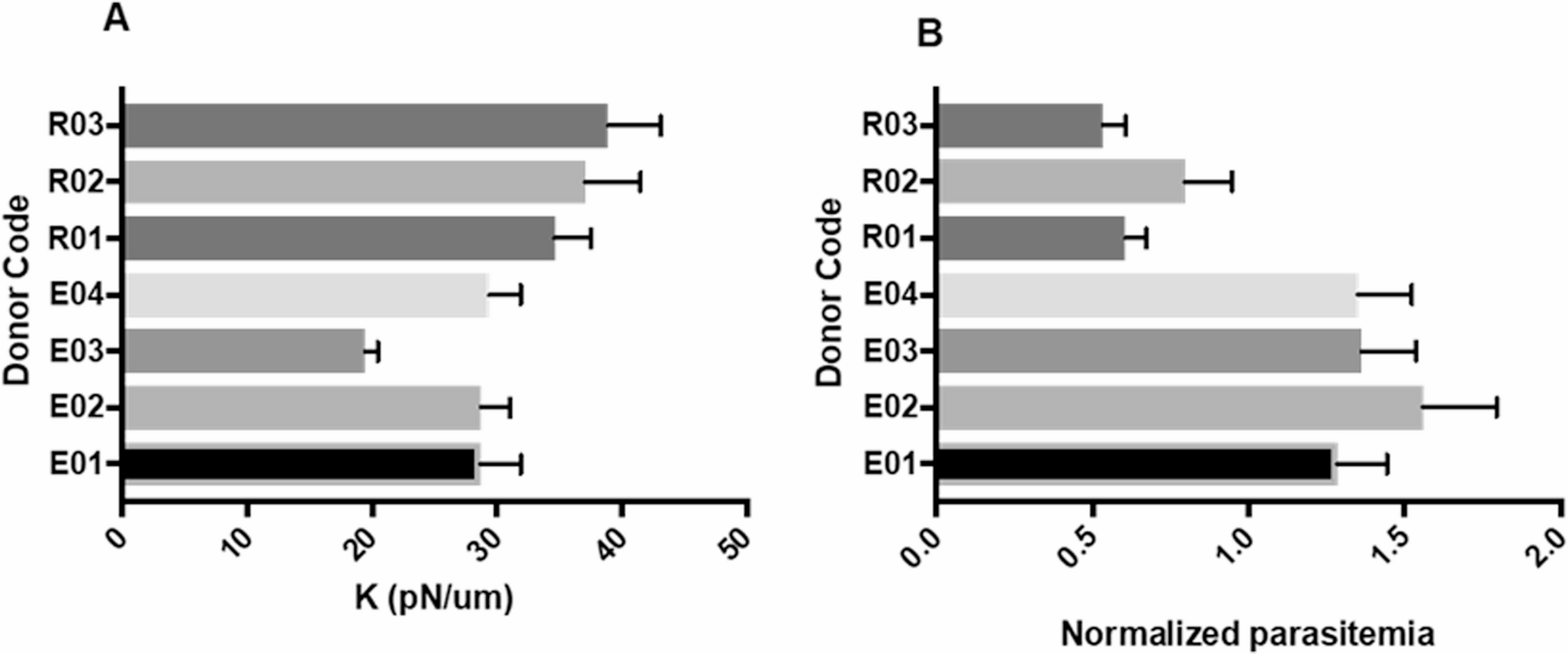
Beyond host genetics, previous studies have characterized red blood cell deformability as a functional biomarker for evaluating antimalarial drug efficacy. Using a multiplexed fluidic plunger system [35], it was assessed the mechanical response of trophozoite-infected erythrocytes after four-hour exposure to twelve different antimalarial agents. Most treatments induced a significant increase in cell stiffness relative to DMSO-treated controls. The optical tweezers research of our group strongly supports these data, demonstrating that the stiffness of RBCs from donors directly dictates the infectivity rate of the parasite in in vitro laboratory cultures. We found that donors previously classified as having reduced parasite invasion had significantly more rigid RBCs, with an average elasticity constant of 36.85 pN/µm. This value is 1.39 times greater than the 26.54 pN/µm average observed in donors with efficient invasion (Fig. 1). These results suggest that RBC stiffness directly impacts parasite infectivity so that the intrinsic differences in the biomechanical properties of RBC donors could impact their susceptibility to parasite infection.
Fig. 1
RBC donor stiffness and invasion. (A) Elasticity constant. (B) Percentage of normalized invasion. E01–04 = efficient donors; R1–R3 = reduced donors
Another recent line of research proposed that extracellular vesicles (EVs) may contribute to preparing uninfected erythrocytes (uRBCs) for Plasmodium falciparum invasion by transporting host and parasite-derived kinases, as well as components of the 20 S proteasome. These vesicles are thought to influence the phosphorylation landscape of cytoskeletal proteins in response to early interactions with merozoite ligands. Importantly, several cytoskeletal proteins that undergo phosphorylation and subsequent loss following reticulocyte-binding protein homolog 5 (RH5) engagement are known substrates of the 20 S proteasome, indicating a coordinated role of proteolysis in modulating red blood cell deformability [36,37,38]. (Fig. 2AB)
Fig. 2
Molecular mechanisms linked to deformability changes during the P. falciparum life cycle. (A) Overview of gliding motility in Plasmodium falciparum sporozoites. Key components, including TRAP, aldolase, actin, and myosin A, form the glideosome, which links external substrates to the parasite cytoskeleton to transmit force. Disruption of the TRAP–aldolase interface impairs motility, making it a candidate for therapeutic intervention. (B) Invasion mechanisms and mechanical remodeling of red blood cells. Receptors linked to cytoskeletal anchor points, including the ankyrin complex, spectrin filaments, AMA1 and RON2, trigger phosphorylation cascades and localized proteolysis that reduce membrane stiffness and favor parasite docking. Extracellular vesicles alter the phosphorylation landscape of cytoskeletal proteins, preconditioning uninfected red cells for invasion. Asexual parasites increase in stiffness as they develop into mature forms until they burst, and the cycle repeats. (C) Developmental and biomechanical remodeling of Plasmodium falciparum gametocytes. Early stages (I–II) retain RBC surface antigens, and the microtubule network exhibits minimal changes in deformability. From stage III, the expression of exported proteins and the microtubule network increases in parallel with stiffness and promotes elongation, contributing to immune evasion and splenic sequestration. At stage V, gametocytes undergo sexual differentiation, depolymerize their microtubule array, and downregulate exported proteins. These changes restore deformability, which allows mature gametocytes to re-enter the peripheral circulation and be taken up by mosquitoes during a blood meal. (D) Signaling model regulating deformability in P. falciparum gametocyte-infected erythrocytes (GIE). In immature stages, low expression of PfPDEδ results in high cAMP and PKA activity. This phosphorylation of STEVOR and host cytoskeletal proteins increases membrane rigidity, which promotes sequestration in the bone marrow and spleen. In mature gametocytes, elevated PfPDEδ expression reduces the level of intracellular cAMP, decreasing PKA activity, and STEVOR phosphorylation decreases membrane rigidity. This enables parasite circulation and mosquito uptake
Moreover, elucidating the impact of host factors, including the metabolic state, oxidative stress, and circadian rhythms, on RBC deformability might highlight critical windows of susceptibility or resistance to parasitic infection. In this context, further exploration of RBC “mechanical memory,” whereby previous mechanical stress influences subsequent cellular responses, presents an intriguing avenue for future research [39].
Mechanical Changes during Different Sexual StagesAlthough most parasites continue the asexual replication cycle within RBCs, a smaller proportion commits to sexual differentiation, forming gametocytes, the transmissible forms of P. falciparum. This process, known as gametocytogenesis, occurs when sexually committed merozoites invade uRBCs and, instead of maturing into a trophozoite and schizont, embarks on an alternative developmental pathway comprising five sequential morphological stages (I–V) over approximately 10 days.
During Plasmodium falciparum gametocyte development, gametocyte-infected erythrocytes (GIEs) exhibit dynamic changes in deformability that are regulated by specific molecular mechanisms crucial for immune evasion and parasite transmission. In immature GIEs (stages I–IV), increased stiffness results from STEVOR proteins interacting with the RBC ankyrin–cytoskeleton complex. This increased cell rigidity facilitates sequestration in the bone marrow for gametocyte maturation. This rigidity is linked to extensive structural reorganization within the parasite cytoskeleton, notably through the expansion of a dense network of microtubules that support the developing gametocyte, defines its polarity, and maintains its cellular shape [11, 40,41,42]. As gametocytes progress to maturity (stage V), intracellular cyclic AMP (cAMP) levels decrease, reducing PKA activity and STEVOR phosphorylation. This shift weakens cytoskeletal interactions, thereby increasing GIE deformability and allowing their release into the peripheral circulation for mosquito uptake circulation and maximizing transmission potential [11, 43,44,45,46,47,48,49]. Flexibility is critical for mature gametocytes to re-enter the peripheral circulation and become available for uptake by mosquitoes during subsequent blood meals. (Fig. 2C) [50, 51].
These finely regulated changes in RBC deformability during gametocytogenesis are not merely structural consequences of parasite development but rather essential adaptive mechanisms that optimize gametocyte maturation, protection, and transmission. Over time, this phenomenon has garnered increasing attention, accompanied by considerable advances in the tools used to study it, from pioneering mechanical deformation methods to cutting-edge techniques involving microfluidics, optical tweezers, and spectroscopic approaches.
Structural targets linked to deformability as potential therapeutic strategies in malaria treatmentRBC deformability represents a critical biophysical parameter underpinning Plasmodium falciparum invasion, immune evasion, persistence, and transmissibility [2, 3, 52]. Recent computational and experimental studies support the existence of an elasticity–rigidity threshold, beyond which excessive membrane tension inhibits the deformation and wrapping processes required for merozoite internalization [13, 53]. This threshold underscores the role of deformability as a physiological prerequisite for successful invasion.
Moreover, several antimalarial compounds affect not only parasite viability but also the mechanical characteristics of infected cells (Table 1). Although these biomechanical alterations may appear indirect, they can have direct consequences for drug efficacy and parasite clearance, especially in P. falciparum strains that are resistant to frontline therapies [54,55,56].
Table 1 Summary of structural targets linked to deformability in iRBCsChanges in cellular stiffness may result from physical disruption of membrane architecture or from interference with signaling pathways that control cytoskeletal remodeling. These effects can compromise parasite development or egress and may help explain the pharmacodynamic differences observed between sensitive and resistant strains. Recognizing and characterizing these deformability-linked effects is thus essential for the rational design of next-generation antimalarials that target not only parasite viability but also the mechanical fitness required for immune evasion and transmission.
A variety of molecular structures—either endogenous to the RBCs or exported by the parasite—are implicated in modulating cell deformability and thus represent potential therapeutic targets. However, the clinical exploitation of these targets is complicated by the structural similarity between host and parasite proteins, raising concerns about their specificity and toxicity [86]. In parallel, the extensive antigenic variation of exported virulence factors such as PfEMP1 and STEVOR limits the feasibility of designing broadly effective interventions [2].
These limitations were revealed in a study by Scovino, in which he described how programmed red blood cell death, a known contributor to red blood cell stiffness that promotes immune clearance of the parasite, exacerbated anemia and vascular dysfunction. This example illustrates the trade-offs involved in targeting host deformability [87].
Recent studies have expanded on this topic, proposing a morphological classification framework to interpret drug-induced phenotypes related to cytoskeletal and membrane alterations. For example, inhibitors of kinases and proteases impair schizont segmentation and egress, producing mechanical phenotypes such as trapped merozoites and asynchronous rupture. Other compounds such as Brefeldin A alter host deformability by disrupting protein export pathways. These phenotypes provide mechanistic insights and suggest the utility of including biomechanical assays in antimalarial compound screening [88, 89].
Collectively, these findings underscore that RBC mechanical properties are dynamically regulated and constitute a viable therapeutic axis. The integration of biophysical assays, molecular profiling, and computational modeling offers a rational pathway for the development of deformability-based antimalarial strategies that selectively counteract parasite adaptations while preserving host cell function.
Deformability as a target in pre-erythrocytic stagesAldolase–TRAP ComplexThe thrombospondin-related anonymous protein (TRAP) is a key micronemal adhesin expressed by Plasmodium sporozoites that is essential for linking extracellular adhesion to intracellular motility via its interaction with aldolase, a glycolytic enzyme that is co-opted as a structural anchor [90,91,92,93,94,95]. In addition to its motility function, TRAP also contributes to the mechanical deformation of host cell membranes, a critical prerequisite for hepatocyte entry and tissue migration [58] (Fig. 2A).
Some small molecules that target the TRAP-aldolase complex have been shown to significantly reduce P. berghei sporozoite motility and hepatocyte invasion. Targeting this complex offers a biomechanically grounded strategy to impair invasion. Given that membrane deformation is mechanically coupled to this complex, its disruption likely impairs the cytoskeletal engagement needed for invasion [59].Notably, a subset of compounds that destabilized the TRAP–aldolase interface paradoxically increased sporozoite invasion in vitro. Although the precise mechanisms remain unclear, this observation suggests a potential mechanosensitive plasticity of the complex or off-target effects that alter its functionality. These nonlinear responses underscore the complexity of biomechanical regulation at the invasion interface and constitute a reason to avoid simplistic interpretations of target inhibition.
Together, these findings position TRAP–aldolase as a structurally and functionally validated modulator of host membrane deformability. Its strategic role in anchoring and gliding initiation makes it a promising target for transmission-blocking therapies aimed at the mechanical interface between the parasite and the host.
Deformability as a target in asexual stagesEBL FamiliesThe erythrocyte-binding-like (EBL) family in P. falciparum, which includes EBA-175, EBA-140, and EBL-1, constitutes a group of Duffy-binding-like (DBL) domain ligands essential for erythrocyte invasion. These proteins selectively recognize sialylated glycophorins, including GPA, GPC, and GPB, on the host membrane, initiating both the adhesion and localized membrane deformation necessary for merozoite internalization (Figure 2B) [102,103,104,105,106,107].
EBA-175, the most extensively characterized member of the Plasmodium falciparum erythrocyte binding antigen (PfEBA) family, binds sialylated O-linked glycans on glycophorin A (GPA), a major erythrocyte membrane glycoprotein anchored to the spectrin–actin skeleton. This interaction initiates cytoskeletal rearrangements that enable membrane wrapping around the invading parasite [61, 99]. Inhibition via synthetic sialic acid analogs or monoclonal antibodies targeting conserved epitopes significantly impairs invasion [63, 64].
Recent findings by [12] identified CD44—a membrane glycoprotein linked to the cytoskeleton—as a coreceptor essential for efficient invasion by P. falciparum. EBA-175–induced phosphorylation of erythrocyte cytoskeletal proteins was shown to be CD44-dependent, linking this coreceptor not only to ligand recognition but also to mechanical remodeling of the host membrane [12, 101]. This mechanism positions CD44 as a critical node in parasite-driven modulation of deformability—an essential biophysical feature for successful internalization. These conserved, essential interactions offer promising targets for antimalarial strategies aimed at minimizing resistance.
EBA-140 recognizes glycophorin C (GPC), a sialylated membrane glycoprotein, through its region II (RII), which is composed of two DBL domains that form distinct glycan-binding sites [68, 102]. Neuraminidase treatment abolishes this interaction, confirming its strict dependence on terminal Neu5Ac residues. GPC is anchored to the cytoskeleton via 4.1R and Ankyrin; therefore, EBA-140 engagement may influence local membrane rigidity, although its precise biomechanical impact remains to be fully characterized [34].
EBL-1 interacts specifically with glycophorin B (GPB), which also bears sialylated O-glycans [66] A functional 69-amino acid segment (F2i) within the DBL domain mediates this binding. Invasion assays confirmed partial inhibition by recombinant F2i peptides, highlighting EBL-1 as a target whose inhibition may stabilize cytoskeletal tension and preserve erythrocyte viscoelasticity during early invasion.
Collectively, the EBL protein family forms a crucial axis of ligand‒receptor interactions that dictates early biomechanical remodeling at the host‒parasite interface. Their reliance on membrane-anchored sialoglycoproteins, which are structurally supported by the erythrocyte skeleton, means that they act not only as invasion ligands but also as active modulators of erythrocyte deformability. Therefore, targeting these interactions could preserve mechanical stability, reduce invasion efficiency, and enhance splenic clearance. This represents a promising therapeutic approach that effectively links molecular specificity with biomechanical intervention.
AMA1-RON2 complexApical membrane antigen 1 (AMA1) is a transmembrane protein of Plasmodium falciparum that is essential for the merozoite invasion of RBCs. AMA1 interacts with rhoptry neck protein 2 (RON2), forming a critical bridge that initiates tight junction formation between the parasite and host cell. This molecular engagement is not only central to receptor recognition but also catalyzes membrane deformation and cytoskeletal remodeling—a mechanical transformation pivotal for parasite entry (Fig. 2B) [116,117,118].
The inhibition of AMA1 through small molecules has been explored by groups that used structural and high-throughput screening approaches to disrupt the AMA1–RON2 interaction, screened over 21,000 small molecules and identified several active candidates. Additionally, an antibody against AMA1 was confirmed to block RBC invasion by merozoites [105].
Other groups have shown promise in the gene-level inhibition of AMA1 using antisense oligonucleotides in rodent malaria models, reinforcing the essential role of AMA1 in invasion and suggesting that transcript-targeting approaches may offer a viable means of biomechanical disruption at the host‒parasite interface [106].
These findings validate the druggability of the AMA1 groove and support its targeting as a strategy to preserve erythrocyte deformability and block parasite entry. This opens a therapeutic avenue focused not only on blocking invasion but also on mitigating the biomechanical consequences of infection.
PfRh4-CR1 and PfRh5-BSGAmong the multiple invasion pathways exploited by Plasmodium falciparum, the PfRh4–CR1 axis is particularly notable for its functional importance and potential biomechanical consequences. PfRh4 mediates sialic acid-independent invasion by binding complement receptor 1 (CR1) on erythrocytes, especially under conditions where sialic acid-dependent ligands such as EBA-175 are inactive or proteolytically cleaved (Fig. 2B) [107]. This interaction promotes tight junction formation, initiating localized cytoskeletal remodeling and membrane curvature, which are essential for merozoite formation [72]. Inhibiting this process could preserve erythrocyte deformability—a critical property for ensuring capillary flow and splenic clearance [108]. Despite its therapeutic potential, no small-molecule inhibitors have been developed for PfRh4, likely owing to challenges in expressing the protein in its native conformation and the intrinsic difficulty of disrupting large protein–protein interfaces with small molecules.
In addition to its mechanical role, PfRh4 contributes to immune evasion through extensive allelic variation. Serological studies in endemic regions have shown that naturally acquired IgG1 and IgG3 antibodies targeting PfRh4 are associated with reduced parasitemia and in vitro inhibition of merozoite invasion, reinforcing its candidacy for multivalent vaccine formulations [109]. Although PfRh4 has not been directly implicated in cytoadhesion phenotypes such as rosetting, its function in tight junction assembly suggests an indirect role in membrane remodeling and potentially in altering erythrocyte biomechanics during invasion. Understanding how immune responses to PfRh4 influence both invasion efficiency and host cell deformability could guide future strategies that integrate immunological and biophysical approaches.
Deformability as a target in sexual stagesSTEVOR, Phosphodiesterase and cAMP Signaling PathwayIn Plasmodium falciparum gametocyte development, phosphodiesterase delta (PfPDEδ) plays a key role in regulating the mechanical properties of infected red blood cells (GIEs), impacting their ability to circulate in the bloodstream and survive. Durin
Comments (0)