
Remember me


You have full access to this article via your institution.
In a recent study, Ghoochani et al. performed a comprehensive quantitative proteomic analysis of the mouse brain to reveal the cell-type-specific composition of lysosomes. The dataset is publicly accessible on Brain LysoAtlas.
Lysosomes are dynamic, membrane-bound organelles that maintain cellular homeostasis by regulating intracellular degradation, signaling, and metabolism.1 Impaired lysosomal protein activity leads to a group of rare inherited conditions collectively known as lysosomal storage disorders (LSDs). Although each LSD is individually rare, together they affect approximately 1 in 5000 individuals. The clinical spectrum is highly variable; however, many LSDs manifest in early childhood and are characterized by progressive neurodegeneration with multisystem involvement. Variants of lysosome-related genes, as well as environmental factors, can also contribute to lysosomal dysfunction, driving neurodegenerative diseases including Parkinson’s disease (PD), Alzheimer’s disease, and related dementias (ADRDs). Despite significant advances in the understanding, diagnosis, and treatment of lysosome-associated diseases, the precise cellular mechanisms driving disease pathology in many of these conditions remain poorly understood.
Characterizing the molecular composition of intact lysosomes is technically challenging, in part because these organelles constitute only a small fraction of total cellular volume. The lysosomal immunoprecipitation (LysoIP) strategy has markedly improved the speed and efficiency of lysosome isolation, initially in cultured cells and mouse tissue.2,3 Applications of LysoIP, as well as variations of this approach, have provided important insights into the molecular basis of lysosomal diseases, including Niemann–Pick type C, underscoring the broad utility and transformative potential of this methodology.4
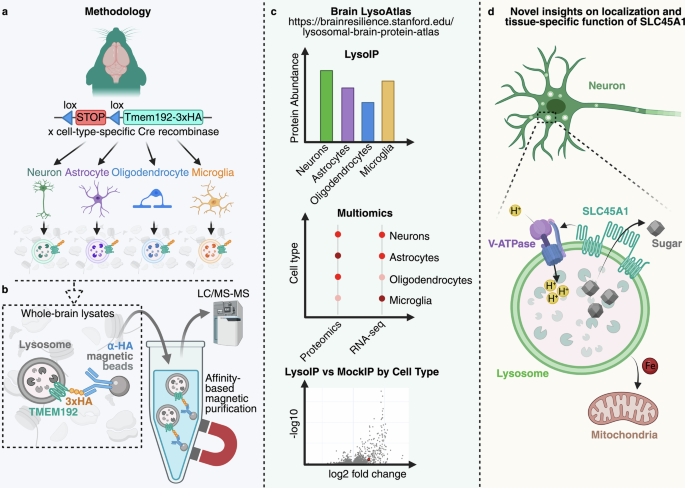
Ghoochani et al. further leveraged LysoIP by crossing LysoTag-lox-stop-lox mice with mice expressing Cre recombinase under different promoters, enabling the purification of intact lysosomes in a cell-type-specific manner from complex and heterogeneous tissues such as the brain5 (Fig. 1a). They combined this approach with the robust sensitivity of label-free, data-independent acquisition (DIA) mass spectrometry to generate a proteomic landscape of lysosomes from mouse neurons, astrocytes, microglia, and oligodendrocytes (Fig. 1b). This quantitative analysis determined protein abundance in each cell type and its enrichment within lysosomes, enabling relative comparisons across cell types. The data are publicly available through a user-friendly interface at https://brainresilience.stanford.edu/lysosomal-brain-protein-atlas (Fig. 1c).
Fig. 1: LysoIP reveals cell-type-specific differences in lysosomal composition in the brain.
a A transgenic mouse model encoding the TMEM192-3×hemagglutinin (HA) fusion protein downstream of a lox-stop-lox cassette was crossed with mice expressing Cre recombinase under cell-type-specific promoters: Syn1 (neurons), Gfap (astrocytes), Olig2 (oligodendrocytes), and Cx3cr1 (microglia). Brains from each line were collected for downstream analyses. b Whole-brain lysates were subjected to affinity-based magnetic purification of lysosomes expressing TMEM192-3×HA. Purified lysosomes were analyzed by label-free, data-independent acquisition liquid chromatography–tandem mass spectrometry (LC–MS/MS). c Mass spectrometry data were used to generate a lysosomal protein atlas across the major brain cell types. d SLC45A1 was identified as a novel neuron-specific lysosomal protein responsible for lysosomal sugar export, vacuolar ATPase (V-ATPase) regulation, and mitochondrial iron homeostasis. Image created using BioRender.
A total of 790 proteins were identified as enriched in purified lysosomes in at least one of the tested brain cell types relative to a mock IP from a mouse not expressing LysoTag. Nearly 22% were enriched in lysosomes across neurons, astrocytes, microglia, and oligodendrocytes. Most of these were previously annotated as lysosomal proteins and included core machinery components such as luminal hydrolases and proteins mediating catabolite export, pH regulation, lysosomal integrity, trafficking, and fusion processes. The abundance of proteins with metabolic functions was even predominantly conserved across cell types. In contrast, the remaining 78% of proteins were not enriched in all datasets. This demonstrates that the abundance of a given protein can differ between cell types or be restricted to specific cell types. Notably, roughly two-thirds of the total proteins identified had not previously been associated with lysosomes. These may represent novel lysosomal machinery, tissue-specific mechanisms, or degradative cargoes.
The authors further demonstrate that this dataset is not merely a resource, but a powerful, successfully applied tool by validating SLC45A1 as a novel neuron-specific lysosomal protein in the brain. SLC45A1 was previously thought to function as a sugar transporter at the plasma membrane. However, extensive analyses demonstrated that SLC45A1 localizes to neuronal lysosomes (Fig. 1d). Cells deficient in SLC45A1 exhibit expansion of the lysosomal compartment, disruption of its ultrastructure, altered autophagy, and reduced proteolytic activity. Furthermore, loss of SLC45A1 leads to hexose accumulation within lysosomes and a significant reduction in vacuolar ATPase (V-ATPase) V1 subunits on the lysosomal membrane. Given that nutrients, including glucose, are known to regulate V-ATPase assembly and activity, the authors speculate that SLC45A1-mediated sugar transport may influence V-ATPase activity and impair lysosomal acidification.
SLC45A1 deficiency also disrupts cellular iron homeostasis, increases ROS levels, and leads to mitochondrial dysfunction — all mechanistic hallmarks of LSDs. Indeed, mutations in SLC45A1 cause a monogenic neurological disorder characterized by epilepsy and intellectual disability.6 Although the brain is affected in most LSDs, these diseases often present as multisystemic conditions and have long been associated with involvement of other organs, including enlargement or dysfunction of the liver and spleen (hepatosplenomegaly), as well as the bone marrow and, in some cases, the lungs. Disorders classified as LSDs have historically been attributed to lysosomal proteins that are ubiquitously expressed.1 Given that SLC45A1 is expressed exclusively in brain neurons, the resulting pathology is restricted to a neuronal phenotype, and a prior diagnosis of LSD was therefore overlooked.5 These findings demonstrate the clinical importance of defining lysosomal composition: the proposed reclassification of SLC45A1 deficiency as an LSD may broaden the scope for diagnosis and facilitate the development of treatment options for current and future patients.
Unlike SLC45A1, most genetic drivers of LSDs, PD, and ADRDs were found to be expressed across multiple cell types. However, differences in expression levels may render certain cell types more susceptible to lysosomal dysfunction, resulting in distinct pathologies. The authors note that it is first necessary to determine whether the difference in abundance scales with the difference in expression, as some proteins not expressed in certain cells were found in their lysosomes. This may be due to protein shedding by one cell followed by uptake by another. This phenomenon is already being exploited in lysosomal enzyme replacement therapy, in which enzymes produced by a subset of cells are released and functionally taken up by deficient cells throughout the tissue.7 However, the precise mechanisms of transfer remain unclear. Lysosomes regulate extracellular vesicle (EV) biogenesis and clearance, and there is growing evidence for EV-mediated transfer of cytosolic or membrane proteins in vivo.8 Lysosomal dysfunction in LSDs, PD, or ADRDs may enhance the release or intercellular transfer of pathogenic proteins or damaged organelles.9 Therefore, current research focuses on modulating lysosomal function or EV secretion and harnessing these pathways to deliver therapeutic enzymes to target cells.
In summary, Ghoochani et al. have established a robust foundation for discovering novel lysosome-related functions and pathologies using Brain LysoAtlas.5 Notably, this group has since published lysosome extraction protocols compatible with downstream liquid chromatography-based metabolomics and lipidomics for future multi-omic analyses.10
Comments (0)