
Remember me

Historically, neutrophils and Th17 cells have been regarded as the primary effector cells in HS [3]. However, B cells are increasingly recognised as immunologically active and pathogenic contributors to its chronic inflammatory milieu. Recent single-cell RNA sequencing and CyTOF analysis by Gudjonsson et al. revealed that B cells and plasma cells are the predominant immune populations in lesional HS skin, outnumbering both T cells and neutrophils [4]. These cells localise within deep dermal niches surrounding epithelialised tunnels, with their abundance strongly correlating with disease severity. Notably, lesional plasma cells expressed tumour necrosis factor (TNF)-α, implicating them in keratinocyte hyperplasia and the perpetuation of inflammation via cytokine-driven amplification loops [5].
Beyond their localisation and abundance, B cells in HS exhibit transcriptional and phenotypic signatures indicative of active participation in disease pathology. Compared to psoriasis and atopic dermatitis, HS displays a unique B cell enriched transcriptional profile marked by the upregulation of CD19, CD79A, and immunoglobulin genes, along with increased frequencies of circulating plasmablasts and CD138⁺ plasma cells in lesional skin [4, 5]. The co-expression of transcription factors IRF4, BLIMP1, and XBP1 further supports in situ differentiation of B cells into antibody-secreting plasma cells [5].
Systemic immune profiling adds further support to this B cell driven paradigm. Flora et al. conducted a proteomic analysis of HS patient serum, revealing enrichment in humoral immune pathways, including IL-13 signalling and immunoglobulin production [6]. Treatment with fostamatinib, a spleen tyrosine kinase (SYK) inhibitor, not only led to clinical improvement in most patients but also reduced serum levels of CCL19 and CCL20, chemokines implicated in recruiting B and T cells to tertiary lymphoid structures (TLSs), particularly among treatment responders.
Taken together, these data highlight the immunologically active and pathogenic role of B cells and plasma cells in HS. Their involvement spans both lesional and systemic compartments, reinforcing the concept of HS as a disease with significant humoral immune dysregulation. These insights support the rationale for targeted therapeutic strategies aimed at modulating B cell function in defined patient subgroups, particularly those with moderate-to-severe or treatment-refractory disease.
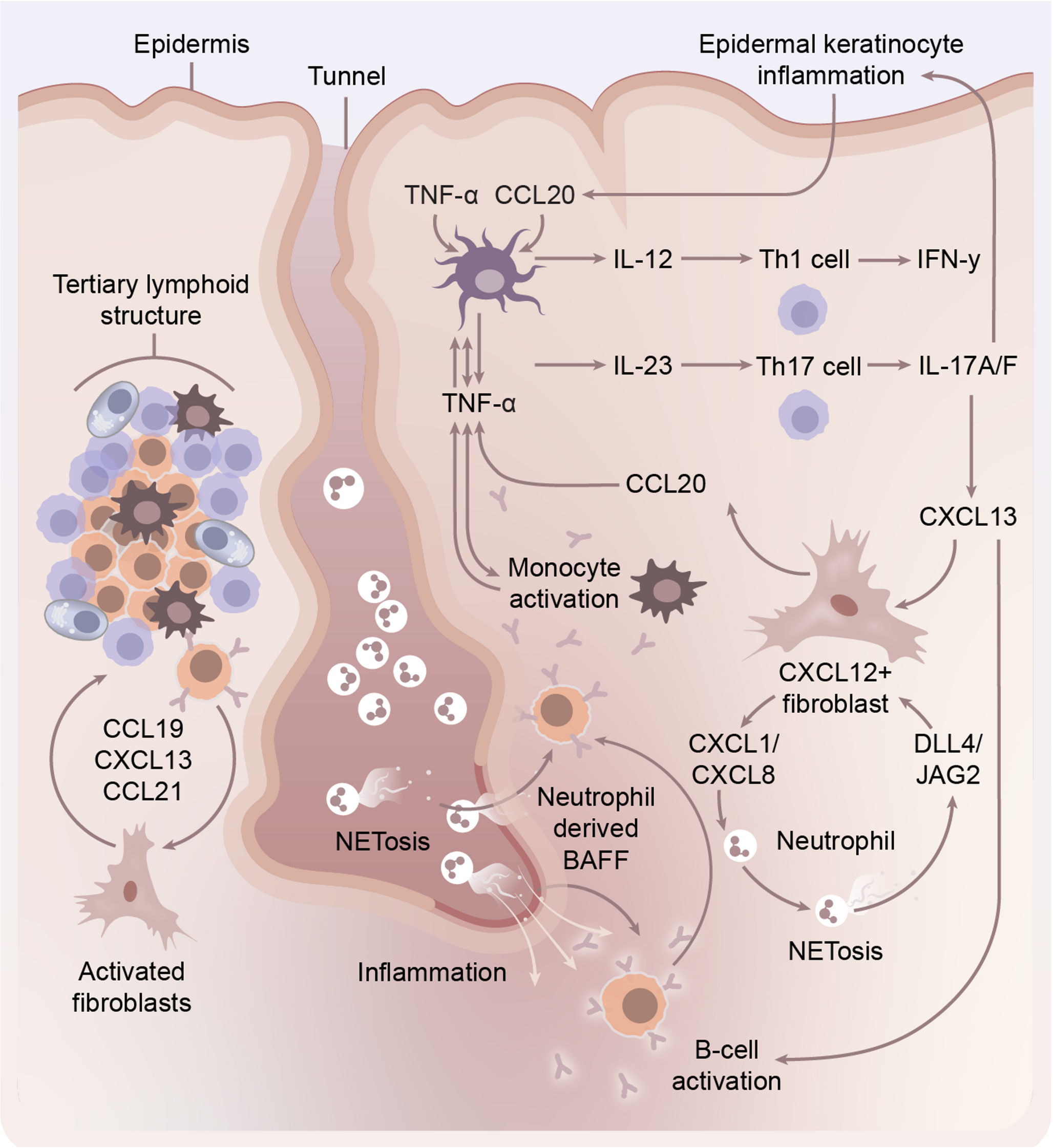
Tertiary Lymphoid Structures and Autoimmunity in HSThe discovery of TLSs in HS has reinforced the central role of adaptive immunity in its pathogenesis. TLSs are ectopic, organised aggregates of immune cells that resemble secondary lymphoid organs (SLOs), forming in response to chronic antigenic stimulation in non-lymphoid tissues [7]. Unlike SLOs, which arise during embryogenesis, TLSs emerge de novo in response to chronic antigenic stimulation and persistent inflammation in non-lymphoid tissues.
TLS development is driven by the coordinated activity of lymphoid tissue inducer (LTi)-like cells, such as activated B cells, T cells, macrophages, and innate lymphoid cells, and lymphoid tissue organiser (LTo) cells, including fibroblasts and endothelial cells [8]. These interactions are mediated primarily through lymphotoxin (LT) and TNF signalling pathways. These pathways upregulate chemokines such as CXCL13, CCL19, and CCL21, which facilitate the recruitment and spatial organisation of lymphocytes into B and T cell zones.(Fig. 1). Expression of activation-induced cytidine deaminase (AID) within TLSs supports local somatic hypermutation, class switching, and germinal centre-like activity, affirming their functional resemblance to SLOs.
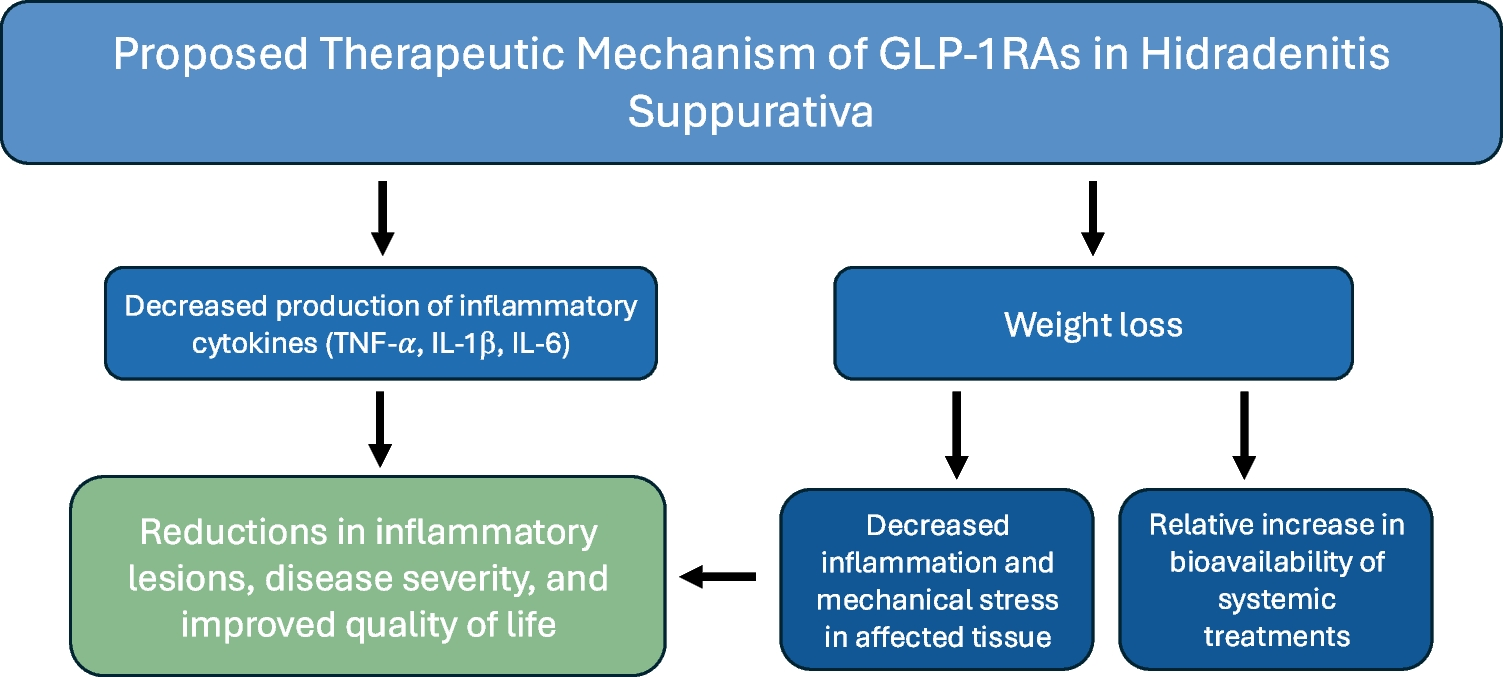
Fig. 1
Schematic Representation of the role of B cells in the pathogenesis of Hidradenitis Suppurativa and interaction with other cellular actors in disease. Tertiary lymphoid structures, in response to chronic antigenic stimulation result in production of chemokines which recruit B lymphocytes to areas of inflammation in HS, primarily around areas of epithelialised tunnel formation. Neutrophils in HS undergoing NETosis release BAFF, activating B cells which in turn stimulate chronic inflammatory pathways through monocyte mediated inflammatory circuits. CXCL12 + fibroblasts are an important inflammatory mediator between B cells and neutrophils via CXCL chemokines production and response to JAG2 and DLL stimulation
Consistent with observations in other autoimmune and chronic inflammatory diseases, TLSs in HS function as hubs of sustained local adaptive immunity. Through spatial transcriptomics and immunofluorescence, Lowe et al. identified TLSs in the deep dermis and subcutis of HS lesions, often in proximity to epithelialised tunnels [9]. These TLSs displayed distinct B and T cell zones, CD21⁺ follicular dendritic cells, and evidence of active germinal centre reactions, including expression of AICDA, MYC, and APEX1, suggesting ongoing affinity maturation and class-switch recombination.
Building on prior observations, Yu et al. identified clonally expanded, somatically mutated IgG1⁺ plasma cells within TLSs in HS lesional skin, which were also detectable in systemic circulation [10]. These plasma cells produced autoantibodies targeting terminally differentiated keratinocytes, with preferential binding to the stratum granulosum and corneum. This was associated with immune complex deposition, complement activation (C4d and C5b-9), and neutrophil infiltration with NET formation, collectively supporting a pathogenic humoral immune response and autoimmune contribution to HS. Fibroblasts isolated from HS tissue expressed CXCL13 and CCL19 in response to TNF-α, IL-1β, and IL-17 A, promoting lymphocyte recruitment and aggregation. Notably, TLS formation was recapitulated in a TLS-on-a-chip model, in which HS fibroblasts induced B and T cell clustering via CXCL13/CCL19 feedback loops. This process was inhibited by early TNF-α blockade but became irreversible once TLSs were fully formed. TLSs were predominantly found in Hurley stage 3 and tunnel-rich disease, suggesting a role in treatment resistance and disease chronicity. Their presence may mark a transition from innate-driven inflammation to chronic autoimmunity and sustained adaptive responses. These findings underscore the importance of early intervention to prevent TLS maturation and highlight fibroblast-immune crosstalk as a promising therapeutic target in severe or refractory HS.
Comments (0)