In this study we describe likely pathogenic or rare missense variants in the BMPR2 pathway gene SMAD6 in 4 of 32 (12.5%) patients with CHD-APAH and in 2 out of 311 (0.64%) patients with IPAH. All of the former patients presented with complex CHD. Four of these in total seven variants were classified as likely pathogenic and could explain at least in part the patients’ phenotype, while three further variants were classified as variants of uncertain significance due to the lack of functional data clarifying the variant’s impact on the SMAD6 protein. One patient had bi-allelic SMAD6 variants. In this patient we were able to identify for the first time all three so far known SMAD6 associated phenotypes in a single individual including radioulnar synostosis, congenital heart defects and craniosynostosis in addition to PAH as a novel SMAD6 phenotype.
In our cohort the sex distribution of SMAD6 variant carriers was equal, even though 81% of the analyzed CHD-APAH and 68% of the IPAH patients were female, in line with previous reports21. In contrast, a male predominance has been observed among rare, deleterious SMAD6 variant carriers with CHD (17:9)13,14,22,23,24,25. Both phenotypes occurring together may, therefore, offer an explanation for the equal sex-distribution of SMAD6 variant carriers in our cohort.
All four CHD-APAH patients with SMAD6 variants had a complex form of heart disease. A common finding for these rather different types of CHD is that the presence of PAH is not obligatory, even if the heart defects are left surgically untreated. It is known since long from a clinical standpoint that only part of these patients is prone to develop PAH, and screening for signs of CHD-APAH has always been prominently implicated in their follow-up. It is unclear why only a subset of patients with the same heart defect goes on to develop PAH. A potential trigger could be a genetic predisposition. To this end, our findings suggest that SMAD6 may be a novel gene associated with CHD-APAH and potentially IPAH.
Previously reported cardiac malformations in SMAD6 variant carriers were either complex cardiac defects and/or characterized by an aortic phenotype13,14,22,24,25. In all three SMAD6 loss-of-function variant carriers in our study an aortic phenotype such as bicuspid aortic valve, aortic endocarditis or aortic stenosis has been identified. Such phenotypes were reported in 10/58 (17%) SMAD6 loss-of-function variant carriers without PAH (Supplementary Table 1). While cardiac malformations have been previously described due to heterozygous pathogenic variants in the SMAD6 gene10,11,12, only a single patient with PAH has been reported up to date14. In addition, it remains unclear why some SMAD6 patients develop craniosynostoses14 and/or radioulnar synostoses15 and others a cardiac phenotype.
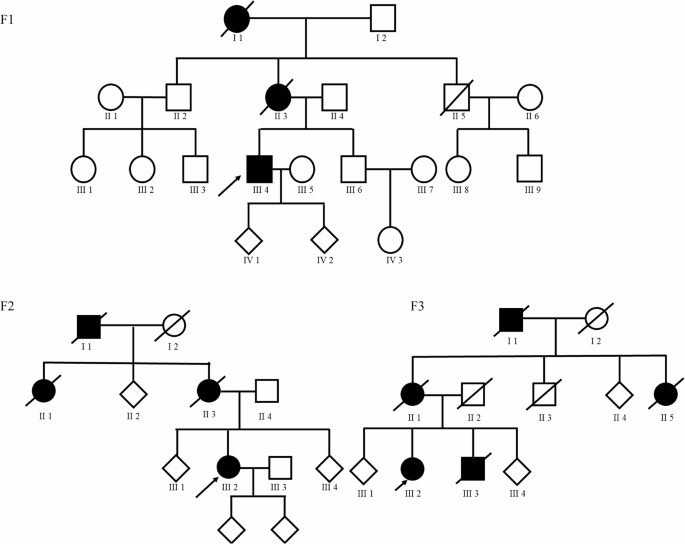
Only our bi-allelic SMAD6 CHD-APAH patient, who was diagnosed at 7 months of age showed all of the other SMAD6 phenotypes. In contrast, the remaining patients in our CHD-APAH cohort were diagnosed solely with congenital heart defects and PAH manifesting at a later stage in life. So far, 4 patients with bi-allelic variants in the SMAD6 gene have been reported, exhibiting either more complex cardiac malformations, or featuring two of the three associated SMAD6 phenotypes22,23.
The other SMAD6 associated disorders are, just as PAH, inherited autosomal dominantly with a reduced penetrance10. This reduced penetrance was particularly evident in the family of the bi-allelic patient. The father of the index patient also carried the same likely pathogenic frameshift variant, yet did not exhibit congenital heart defects nor develop PAH. However, a slightly elevated systolic PAP during exercise echocardiography could be observed. Moreover, as he passed away at a young age due to an accident. PAH may still have manifested later in his life and it is inconclusive whether he had a higher vulnerability to cardiac conditions.
Retrospectively, the patient with IPAH and comorbidities and a likely pathogenic SMAD6 frameshift variant displayed a susceptibility to cardiac and vascular events with an aortic valve endocarditis, aneurysm and dissection in the right groin. Interestingly, not only this but also four other SMAD6 variants described in this study have been reported previously in patients with aortic valve disease albeit without PAH (ClinVar database, Table 4).
SMAD6, like SMAD7, is an inhibitory SMAD protein of the TGF-β superfamily. It predominantly targets the BMPR2 signaling pathway26,27. In the presence of abundant growth factors, SMAD6 is increasingly synthesized and acts as a negative feedback molecule28.
SMAD6 exhibits its inhibitory properties by several mechanisms. Firstly, it can bind to the BMP type 1 receptor, thereby preventing the phosphorylation of the receptor-related SMADs and inhibiting BMPR2 downstream signaling26. Secondly, SMAD6 recruits the ubiquitin ligase Smurf1 to the BMP type 1 receptor and facilitates receptor ubiquitination and subsequent degradation29. Thirdly, SMAD6 acts via non-canonical pathways such as the Notch signaling pathway30. SMAD6 is downstream of the mechanosensitive Notch 1 pathway, responsible for the flow-mediated cell alignment and barrier function of endothelial cells30. It is hypothesized that vascular remodeling in PAH is attributed, among other factors, to compromised endothelial barrier integrity and dysregulated endothelial proliferation31. The SMAD6 variants could have therefore altered endothelial cell homeostasis and function.
Both, the TGF-β and the Notch signaling pathway, are involved in endothelial-to-mesenchymal transition (EndMT)32. Silencing of SMAD6 favors the development of a mesenchymal phenotype via the unimpaired BMP signaling pathway33. During EndMT, endothelial cells undergo a phenotypic shift, losing their characteristic features and acquiring mesenchymal traits. This process is believed to be implicated in various pathological conditions, including pulmonary hypertension34,35. Thus, a disturbed SMAD6 protein may drive mesenchymal cell features of the endothelium.
During embryonal development SMAD6 plays a key role in the endocardial cushion transformation36. Homozygous Smad6 knockout mice exhibited higher lethality and various cardiac malformations, including valve hyperplasia and septation defects37. Surviving animals presented with aortic ossification, reduced vasodilation, and systemic hypertension. There was overexpression of mesenchymal cells in the heart valves, which could have resulted from an enhanced endocardial to mesenchymal transformation or increased cell proliferation according to Galvin and colleagues37.
Another potential mechanism of PAH development in SMAD6 patients could be the presence of the underlying cardiac defects themselves. Two of our patients with SMAD6 variants exhibited a rare segmental PAH characterized by an abnormal development of pulmonary vasculature due to atresia or agenesis of the pulmonary arteries. None of the patients with SMAD6 variants had a simple heart defect as mainly reported for patients with pathogenic SOX17 or TBX4 variants. In contrast, all heart defects were of moderate to great complexity according to American Heart Association and American College of Cardiology guidelines38. Thus, the heart defect itself could have been the primary cause of PAH. However, not all patients with this phenotype develop PAH. Pathogenic SMAD6 variants could, therefore, be an additional trigger for PAH manifestation offsetting the balance of the TGF-β pathway and the tightly controlled system of tissue homeostasis governed by proliferation and apoptosis.
While a great tolerance to frameshift variants in SMAD6 exists in the general population (pLI score of 0), loss-of-function variants were significantly enriched in our CHD-APAH cohort and missense variants showed a trend of higher prevalence in our CHD-APAH cohort compared to the general population. An overrepresentation of rare missense and protein truncating SMAD6 variants was also reported for CHD patients without PAH, in particular in those with bicuspid aortic valve and thoracic aortic aneurysms10. The elevated frequency of loss-of-function variants also in controls, may have obscured the detection of SMAD6 as a potential novel CHD-APAH gene with reduced penetrance in former case-control studies.
Detailed clinical data and DNA samples from all index patients’ first-degree family members would have been very useful to perform further co-segregation analysis. Apart from the family of patient 2 no further DNA samples were available. From the recorded clinical family data, no further family members showed any signs of PAH or SMAD6 associated phenotypes. Nevertheless, the presence of the SMAD6 variants cannot be ruled out also within families 1 and 3–6 due to the reduced penetrance.
The prevalence of likely pathogenic SMAD6 variants in our CHD-APAH patient cohort (6.3%) was higher than the reported frequency for SOX17 (3.2%) or TBX4 (2.6%) in CHD-APAH patients16. In contrast, SMAD6 variants were not overrepresented in IPAH patients with or without comorbidities. Thus, it is unclear whether SMAD6 variants also contributed to PAH development in the cohort of IPAH patients. Their classification as likely pathogenic does, however, offer at least some support for their disease contribution.
In summary, this is the first study to report SMAD6 as a potentially novel IPAH and CHD-APAH gene. Further studies are necessary to confirm our findings and to determine the exact prevalence of disease-associated SMAD6 variants. We advocate for testing SMAD6 variants in PAH patients, particularly those with complex congenital heart defects or unclear cardiovascular abnormalities, as well as PAH patients with craniosynostosis or radial synostosis. Implementing SMAD6 onto an existing PAH gene panel provides a feasible approach to routinely and cost-effectively screen for pathogenic SMAD6 variants in patients with (CHD-A)PAH. Moreover, testing for SMAD6 variants may positively influence patient management by enabling early detection and treatment of complications such as aneurysms. Similarly, CHD patients with a confirmed (likely) pathogenic SMAD6 variant should be clinically evaluated for PAH aiming for early diagnosis and guideline-adherent management.



Comments (0)