
Remember me
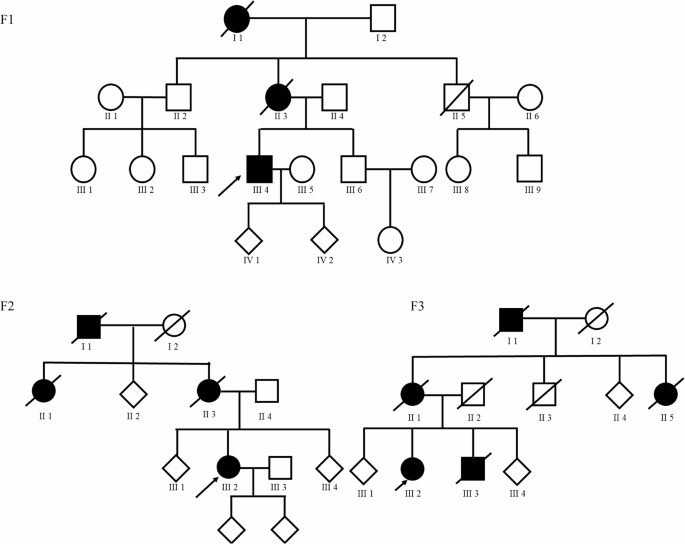


We identified a previously published heterozygous missense mutation, c.212 G > A, p.G71E (NM_004082.5), in exon 2 of the DCTN1 gene in three unrelated PS families (Fig. 1)10. These three probands had the mean age of 47.67 ± 4.04 years and mean age at onset (AAO) of 43.00 ± 4.00 years. The variants of the three probands had been verified by Sanger sequencing, and the results were shown in Supplementary Fig. 1. Detailed clinical manifestations of probands and relevant findings of the affected individuals were provided in Table 1.
Fig. 1
Pedigrees of patients with Perry syndrome. Arrow, point to the proband; square, man; circle, woman; Diamonds, disguised gender; slashed symbol, deceased; filled symbol, affected; empty symbol, unaffected. The figure was created by Adobe Illustrator 2022.
Table 1 Clinical characteristics of Perry syndrome patientsFamily 1: the pedigree contained 20 members including the spouses, spanning four generations, with three affected individuals (Fig. 1. F1). The proband (III-4), who was diagnosed with definite PS, began with bradykinesia, postural instability, and backward falls at the age of 48-year-old. As the disease progressed, he displayed cognitive impairment, reduced facial expression, staring off, slow speech with dysarthria, and difficulty falling asleep. Over the next year, he developed apathy, and severe depression with suicidal ideation and he continued to lose approximately 10 kg in weight. His caregivers subsequently noted that he had an abnormal breathing pattern with rapid shallow breathing. At age 49, his neurological examination revealed predominant axial akinetic-rigid Parkinsonism with diminished postural reflexes. Examination of gaze showed impaired vertical pursuits. The brain MRI had no obvious abnormalities. The proband was unable to finish respiratory function tests and a battery of neuropsychological tests, including the Mini-Mental State Examination (MMSE), Montreal Cognitive Assessment (MoCA), and Hamilton Anxiety Scale (HAMA). The treatment with L-Dopa (levodopa equivalent dose 137.5 mg/d) resulted in no motor gains. His mother (II-3) suffered from cognitive deficits and upper limb tremor, and she died four years after the onset. His grandmother (I-1) committed suicide when she was 40 years old (details unavailable).
The DCTN1 mutation segregated with the disease: patient III-4 carried the mutation, whereas 2 asymptomatic relatives II-2 (older than 60 years) and III-6 (40-year-old), did not (supplementary Fig. 2). The DNA of patients II-3 and I-1, who were putative carriers, was unavailable. In addition, the proband’s father (II-4) declined to provide a blood sample for genetic testing.
Family 2: a forty-year-old right-handed woman reported a 1-year history of progressive slowed movements, imbalance, and anxiety (Fig. 1. F2). She had no anosmia, autonomic disturbances, hallucinations, or vivid dreams, but difficulty in getting to sleep. Examination revealed hypomimia, bradykinesia, reduced arm swing, rigidity in all limbs, dysarthria, and normal cognition (MMSE score 28/30). Her motor symptoms were bilateral and worse on the right. The presynaptic dopamine transporter brain PET imaging showed asymmetric reduced uptake in the basal ganglia (caudate nucleus and putamen), more marked on the left. The clinicians started levodopa, which improved her Parkinsonism partially. In the most recent follow-up assessment after two years from onset, she had developed marked end-of-dose wearing off spells, with the Hoehn-Yahr (H&Y) stage being 3.0, and yet no hypoventilation or weight loss.
Genetic testing revealed the c.212 G > A, p.G71E mutation in exon 2 of the DCTN1 gene in the proband III-2. Her mother, grandfather, and aunt all presented with uniform phenotypes and died of the disease at the ages of 54, 50, and 55. However, detailed information on their disease courses was inaccessible, and these affected members did not seek medical diagnosis or treatment. Meanwhile, the DNA of II-3 and II-4 was unavailable. The proband’s two children were well. Collectively, we diagnosed her with definite PS.
Family 3: a 48-year-old female was in good health until the age of 43 (Fig. 1. F3), when she developed evident Parkinsonism (bradykinesia and asymmetrical rest tremor combining postural tremor, worse on the right side, lead-pipe rigidity in all limbs), depression, sleep difficulties, daytime somnolence, and autonomic dysfunction. She had a masked face, and the MMSE score was 23/30. She had felt unsteady while resting and asleep, with a very strong urge to move her legs. The pedigree indicated an autosomal dominant inheritance: her mother and aunt displayed similar symptoms, and both passed away in their fifth decade of life, but we were unable to gain any detailed information about these two affected family members. Her 43-year-old sibling had complained of upper limb tremors and slowed movements for two years. In the latest follow-up visit, the proband died at the age of 50, while her sibling died at the age of 46. Genetic tests were negative for any known pathogenic mutation of PD, but positive for DCTN1, p.G71E mutation. She fulfilled the definite criteria for diagnosing PS.
Identification of DCTN1 variants in PD cohortIn the EoPD & familial PD (FPD) cohort, 2082 PD patients, including 1593 EoPD patients and 489 FPD patients had the mean age of 52.44 ± 8.67 years and mean AAO of 46.93 ± 8.25 years. In the late-onset PD (LoPD) cohort, the mean age was 66.76 ± 7.08 years and the AAO was 61.88 ± 6.93 years for 1962 LoPD patients (Table 2).
Table 2 Basic demographic characteristics of included subjectsOverall, 68 cases were identified as carriers of rare nonsynonymous variants in the DCTN1 gene, with 30 cases harboring PDVs (28 rare damaging missense (Dmis) variants and 2 loss of function (LoF) variants) (Supplementary Table 1) (Fig. 2). In gene burden analysis, rare PDVs (Dmis plus LoF variants) in the DCTN1 demonstrated a potential association with total PD patients (30 vs. 11, p = 0.042) that was no longer significant after FDR correction (p* = 0.084) (Table 3, Fig. 3). We also performed exploratory subgroup analyses within the PD cohort, which included the EoPD&FPD and LoPD subgroups. A nominally significant association of DCTN1 PDVs with PD was found in the EoPD & FPD cohort (20 vs. 7, p = 0.049), which did not remain significant after correction (FDR p* = 0.098). No such association was detected in the LoPD cohort (10 vs. 4, p = 0.422, p* = 0.422).(Table 3, Fig. 3). Given that mutations within distinct domains are likely to be related to different molecular events through a varied protein interactome, we further investigated the associations of rare PDVs located in different functional domains of DCTN1 with PD risk (CAP domain, CC1 domain, and CC2 domain, respectively). However, no correlations were observed in our analysis (p = 0.191 for CAP-domain, p = 0.118 for CC1 domain, p = 0.941 for CC2 domain) (Supplementary Table 2). In addition, we performed a burden analysis on synonymous variants in the PD cohort. The result demonstrated that synonymous variants in the DCTN1 were not associated with the risk of PD in any of the analyzed cohorts (Supplementary Table 3). Additionally, all variants predicted as PDVs have been verified by Sanger sequencing, and the results are shown in Supplementary Fig. 1. In the UKB cohort, burden analysis at the entire gene level did not reach significant level. (Supplementary Table 4)
Fig. 2: Schematic representation of the DCTN1 protein with variants identified in DCTN1-related disorders.
Variants identified in our cohorts are depicted below the protein schematic: Variants identified in Parkinson’s disease cohort and Perry syndrome patients are listed above the schematic and variants identified in Amyotrophic lateral sclerosis cohort are listed below the schematic. CAP: cytoskeleton-associated protein and glycine-rich domain (amino acids 29–95); Basic: basic domain (amino acids 96-150); CC1: coiled coil domain 1 (amino acids 217–548); CC2: coiled coil domain 2 (amino acids 926–1049); ICD: inter coiled domain (amino acids 549-926); C-ter: C-termini (amino acids 1050-1278); MT binding: microtubule binding (amino acids 39-150); Dynein binding (amino acids 200-811). The protein domain structure was created by Illustrator for Biological Sequences 2.056.
Fig. 3
Forest Plot of Odds Ratios with 95% CIs from Burden Tests Across Parkinson’s Disease and Amyotrophic lateral sclerosis.
Table 3 Analysis of DCTN1 gene rare variant burden at total gene level in Parkinson’s disease and Amyotrophic lateral sclerosisParticularly, 2 PDVs in exon 2 of DCTN1 (namely p.K56R and p.G71E, both documented point mutations in PS) were identified in our EoPD & FPD cohort. We then performed follow-up phone calls on these two patients. Except for the known Parkinsonism manifestations, we carefully inquired if they developed any additional PS-related symptoms, including apathy or depression, respiratory problems, unexplained weight loss, or rapid disease progression within 5 years of onset. We also verified whether they had a positive family history of Parkinsonism or respiratory symptoms. After thorough questioning, we confirmed that they still did not present any of the other core symptoms of PS above; hence, we diagnosed them with PD at this time. This may be owing to the incomplete penetrance of the DCTN1 p.K56R and p.G71E mutations, or it could be because the clinical symptoms have not yet fully emerged, needing long-term follow-up for a definitive final diagnosis. Detailed clinical characteristics in PD patients with DCTN1 PDVs were shown in Supplementary Table 5. Furthermore, we performed genotype-phenotype association analyses to investigate potential modifying effects of DCTN1 PDVs on PD phenotypes. However, our results did not demonstrate any statistically significant association between DCTN1 PDVs and clinical phenotypic manifestations in PD (Supplementary Table 6).
Identification of DCTN1 variants in ALS cohortsWe analyzed the rare variants of DCTN1 gene in 988 ALS patients of Chinese ancestry. A total of 22 rare variants (minor allele frequency, MAF < 0.001) were identified, including 4 splicing site variants and 18 missense variants, among which 10 were predicted as PDVs by Reve. Unlike PS, we found none of these 10 PDVs are inside exon 2. Instead, they are dispersed throughout exons 14 to 26. PDVs at the gene level were not enriched in ALS patients (10 vs 7, p = 0.089, p* = 0.119) (Table 3, Fig. 3). The location and pathogenicity of the 10 PDVs identified in this study are shown in Fig. 2 and Supplementary Table 7. Equally, all variants predicted as PDVs identified in ALS cohort have been verified by sanger sequencing, and the results are shown in Supplementary Fig. 1. In addition, burden analysis was also performed in the Project MinE cohort. A total of 91 rare variants (MAF < 0.001) were identified, among which 28 were predicted as PDVs by Reve. The burden analysis was performed (Supplementary Table 4) to further indicate that DCTN1 may not be an ALS risk gene.
To explore the genotype-phenotype associations, we comprehensively reviewed the clinical manifestations of these ten unrelated ALS patients with DCTN1 PDVs. All these ten patients carrying DCTN1 PDVs were sporadic, with six having spinal onset, three presenting bulbar onset and the remaining one exhibiting multifocal onset of both spinal and bulbar. The mean AAO was 55.40 ± 10.88 years old, with a sex ratio of 1.5:1 (6 men: 4 women). By the last follow-up, 6 patients had died, 1 were lost to follow-up, and 3 were still alive. The median survival time was 27.00 ± 13.97 months. According to the Edinburgh Cognitive and Behavior ALS Screen (ECAS) and MMSE scores, about 60.0% (6/10) of the patients had cognitive impairment. Compared to the other ALS patients in our cohort who did not have DCTN1 PDVs, there were no significant differences in the AAO (55.40 years vs. 54.11 years), proportion of male involvement (40.00 vs 35.48%), or the revised version of the ALS functional rating scale (ALSFRS-R) scores. No specific clinical characteristics were observed in ALS patients with DCTN1 PDVs, detailed in Supplementary Tables 8 and 9.
Comments (0)