
Remember me




Periodontal diseases are chronic inflammatory conditions influenced by the host immune response and alterations in the oral microflora [27,28,29]. These diseases are primarily initiated by biofilms containing pathogenic bacteria that evade the host's defense mechanisms, leading to inflammation and tissue destruction [30]. The pathogenesis of periodontal disease encompasses both local soft tissue pathology and systemic effects resulting from the dissemination of microorganisms and modulation of the host immune response [27, 31, 32].
Role of biofilms and key pathogensDental biofilms are structured communities of microorganisms embedded in an extracellular matrix that adheres to tooth surfaces [33]. This biofilm structure provides bacteria with protection against host immune responses and antimicrobial agents, enabling their persistence and contribution to disease processes [34, 35]. Microbial dysbiosis, a central element in periodontal disease, refers to an imbalance or disruption in the composition and function of the microbiome. Crucially, it is characterized by a shift not merely in the types of bacteria present but fundamentally in their relative abundance, overall community structure, and collective functional output (e.g., altered metabolic activity and increased virulence factor expression). This shift moves the subgingival environment from a health-associated state, typically dominated by commensal Gram-positive organisms, towards a disease-associated state characterized by a predominance of Gram-negative anaerobes. These pathogens release virulence factors, including toxins, that trigger and intensify inflammatory responses [36].
While microbial dysbiosis is central to periodontal disease, understanding the specific changes within the oral microbiome is critical. In a healthy oral cavity, commensal species, such as Streptococcus and Actinomyces, dominate and maintain homeostasis by modulating the immune response and outcompeting harmful pathogens [10]. In contrast, periodontal disease is marked by the proliferation of pathogenic species like Porphyromonas gingivalis, Treponema denticola, and Fusobacterium nucleatum, which thrive under these dysbiotic conditions characterized by altered community dynamics and function [9].
Microbiome variability among individuals significantly influences susceptibility to and progression of periodontal disease. Factors such as genetics, diet, smoking, and systemic health play a vital role in shaping the microbiome's composition and function, resulting in unique microbial profiles [37]. For instance, smokers tend to harbor higher proportions of Treponema and Prevotella species, correlating with increased disease severity [38]. Furthermore, the concept of a "healthy microbiome" is complex, as variations can be observed even among periodontally healthy individuals [29, 39]. Future research aimed at defining microbial signatures of health and disease, considering both composition and functional shifts, is critical for identifying reliable biomarkers and developing personalized diagnostic and therapeutic approaches.
Porphyromonas gingivalisP. gingivalis is a gram-negative, anaerobic bacterium which disturbs the balance of the biofilm community and contributes to bacterial dysbiosis, thus leading to the progression of periodontal disease. Some of its virulence factors include gingipains (cysteine proteases), which digest host proteins, compromise the immune system, and promote tissue invasion [9].
It expresses a weakly immunogenic lipopolysaccharide (LPS), modifies Toll-like receptor (TLR) signaling and hampers neutrophil killing [40].
Treponema denticolaT. denticola is a Gram-negative, obligate anaerobic motile spirochete that is associated with late-stage periodontitis. It forms biofilms that protect the bacteria from the host immune response and antimicrobial treatments. Its dentilisin protease degrades the host extracellular matrix proteins and complements the proteolytic activity of P. gingivalis [41].
This pathogen disrupts epithelial barriers and enables other microorganisms to infect connective tissues [42].
Fusobacterium nucleatumF. nucleatum is another Gram-negative, anaerobic bacterium that acts as a bridge organism in biofilm contributing to the colonization of other pathogens. It adheres to epithelial cells and promotes inflammation by inducing secretion of interleukin-8 (IL-8) and other proinflammatory cytokines [14].
It can invade tissues and bloodstream and has been linked to diseases such as colorectal cancer and adverse pregnancy outcomes [17].
The virulence mechanisms of key pathogens reveal critical insights into periodontal disease progression. Porphyromonas gingivalis employs gingipains to degrade host proteins and modulate the complement system, evading immune detection while promoting inflammation and deeper tissue invasion [9]. Similarly, Treponema denticola utilizes dentilisin protease to break down extracellular matrix components and disrupt cell–cell junctions, potentiating the pathogenicity of the biofilm [41]. Fusobacterium nucleatum excels in adhesion, with outer membrane adhesins enabling its integration into multispecies biofilms and facilitating colonization of diverse niches [17]. These properties underscore its implication in both local and systemic diseases. Improving our understanding of the oral microbiome's complexity and the specific roles of pathogenic bacteria will help to identify targeted therapies and to develop strategies to restore microbial balance. Future research on microbial interactions and host–pathogen dynamics will further refine periodontal diagnostics and treatment approaches.
Mechanisms linking oral and systemic healthPeriodontal pathogens, such as Aggregatibacter actinomycetemcomitans (Aa), Porphyromonas gingivalis (Pg), Tannerella forsythia (Tf), Treponema denticola (Td), Eubacterium nodatum (En), Fusobacterium nucleatum (Fn), Prevotella intermedia (Pi), Campylobacter rectus (Cr), Peptostreptococcus (Micromonas) micros (Pm), Eikenella corrodens (Ec), Prevotella nigrescens (Pn), and Capnocytophaga species (gingavalis, ochracea, sputigena) (Cs), can enter the bloodstream through bacteremia caused by routine activities like chewing, brushing, flossing, and invasive dental procedures [43]. While such bacteremia is typically transient and low-level in individuals with healthy periodontal tissues, efficiently cleared by the host immune system, the situation can be significantly different in periodontitis. In patients with periodontal disease, the inflamed and ulcerated gingival epithelium provides a more persistent portal of entry, and the higher subgingival bacterial load can lead to bacteremia that is more frequent, involves a greater number of microorganisms, and may persist for longer periods [15]. Once in circulation, these pathogens disseminate to tissues and organs, potentially contributing to the pathogenesis of systemic diseases. For instance, the DNA of Pg and Fn has been identified in atherosclerotic plaques, supporting the hypothesis that oral pathogens are linked to cardiovascular diseases. These findings underscore the ability of periodontal bacteria to spread from the oral cavity to other parts of the body, emphasizing the importance of oral hygiene in preventing systemic diseases [44]. These findings underscore the ability of periodontal bacteria to spread from the oral cavity, particularly in the context of disease, emphasizing the importance of oral hygiene and periodontal health in preventing systemic complications.
Periodontal inflammation contributes to systemic diseases through the release of pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, as well as acute-phase proteins like CRP. These inflammatory mediators exacerbate systemic conditions, including cardiovascular diseases, insulin resistance in diabetes, and adverse pregnancy outcomes [12, 20, 45]. LPS, a major outer membrane component of Gram-negative bacteria, is a key driver of this inflammation. However, the immunostimulatory potential of LPS varies significantly depending on its structure, particularly the lipid A moiety, which interacts with the host Toll-like receptor 4 (TLR4) complex. While classical LPS from bacteria like E. coli possesses a highly pro-inflammatory hexa-acylated lipid A structure, periodontal pathogens such as P. gingivalis produce heterogeneous lipid A forms (often tetra- and penta-acylated) [40, 46]. This structural difference renders P. gingivalis LPS a weak agonist or even an antagonist of human TLR4 [40]. This unique characteristic allows P. gingivalis to potentially subvert or modulate the initial strong inflammatory response typically triggered by LPS, contributing to immune evasion and the establishment of chronic, persistent inflammation rather than acute clearance [46, 47]. LPS released by Gram-negative periodontal pathogens, such as Pg, induce endothelial dysfunction, a key event in atherogenesis. LPS activates endothelial cells to express adhesion molecules like E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1), facilitating leukocyte adhesion and migration, which promotes vascular inflammation and plaque formation [27, 48]. Additionally, LPS can disrupt the placental barrier, increasing the risk of preterm births and adverse pregnancy outcomes [16, 49]. Pro-inflammatory cytokines released during periodontal inflammation also play a significant role in systemic metabolic dysregulation. TNF-α inhibits insulin receptor signaling by increasing serine phosphorylation of insulin receptor substrates, impairing glucose uptake in peripheral tissues. Similarly, IL-6 contributes to insulin resistance by enhancing hepatic glucose production and reducing adiponectin levels, an anti-inflammatory cytokine critical for maintaining insulin sensitivity [50, 51]. These interconnected pathways link periodontal inflammation to systemic diseases like diabetes and cardiovascular conditions.
Systemic conditions can also predispose individuals to periodontal disease through mechanisms such as altered immune responses and metabolic dysregulation. For example, diabetes mellitus, particularly when poorly controlled, leads to hyperglycemia-induced impairment of neutrophil chemotaxis and phagocytosis, reducing the host’s ability to combat infection by periodontal pathogens [20, 52]. Elevated glucose levels in gingival crevicular fluid (GCF) create an environment conducive to the growth of pathogenic bacteria, exacerbating periodontal inflammation [53]. Similarly, rheumatoid arthritis, an autoimmune condition, promotes systemic inflammation that weakens the gingival connective tissues and facilitates microbial colonization [54]. Cardiovascular diseases, characterized by chronic low-grade inflammation, may contribute to an inflammatory milieu that accelerates periodontal tissue destruction [55]. Additionally, medications for systemic conditions, such as calcium channel blockers or immunosuppressants, increase susceptibility to periodontal disease by causing gingival overgrowth or dampening immune responses [56].
The host immune response further drives the systemic impact of periodontal disease. Innate immune cells, such as neutrophils, macrophages, and dendritic cells, recognize periodontal pathogens via pattern recognition receptors (PRRs), including Toll-like receptors (TLRs). This triggers the production of pro-inflammatory cytokines, leading to the recruitment of adaptive immune cells like T-helper cells (Th1, Th2, Th17) and B lymphocyte cells. These cells contribute to chronic inflammation and tissue destruction by releasing receptor activator of nuclear factor-kappa B ligand, a protein that promotes osteoclast-mediated bone resorption [10, 47]. Genetic predispositions, such as polymorphisms in cytokine-encoding genes (e.g., IL-1β, TNF-α), heighten inflammatory responses, increasing susceptibility to periodontal and systemic diseases [57]. Molecular mimicry represents another mechanism linking periodontal and systemic health. This occurs when microbial antigens share structural similarities with host proteins, potentially triggering autoimmune responses where the immune system mistakenly attacks host tissues. For example, bacterial Heat Shock Proteins (HSPs), such as HSP60 (GroEL) produced by pathogens like P. gingivalis under stress conditions, exhibit sequence homology with human HSP60. An immune response initially mounted against these bacterial HSPs may subsequently cross-react with human HSPs expressed on host cells, such as endothelial cells in blood vessels (implicated in atherosclerosis) or synovial cells in joints (implicated in rheumatoid arthritis), thereby contributing to inflammation and tissue damage in these systemic conditions [56, 57]. Additionally, virulence factors like gingipains produced by Pg can degrade complement factors, impairing immune clearance mechanisms and perpetuating chronic inflammation, which further amplifies systemic disease progression [46, 58]. Although a growing body of evidence supports associations between periodontal disease and systemic conditions, establishing causation remains challenging [59]. Observational studies demonstrate significant correlations between periodontitis and conditions like cardiovascular disease, diabetes, and adverse pregnancy outcomes; however, confounding factors, such as smoking, socioeconomic status, and comorbidities, complicate these relationships [8, 60]. Reverse causation is also a concern, as systemic diseases like diabetes can exacerbate periodontal conditions through altered immune responses and metabolic dysfunction [12]. Longitudinal interventional studies are essential to address these challenges and clarify whether effective periodontal treatment can mitigate systemic disease risks and vice versa [61].
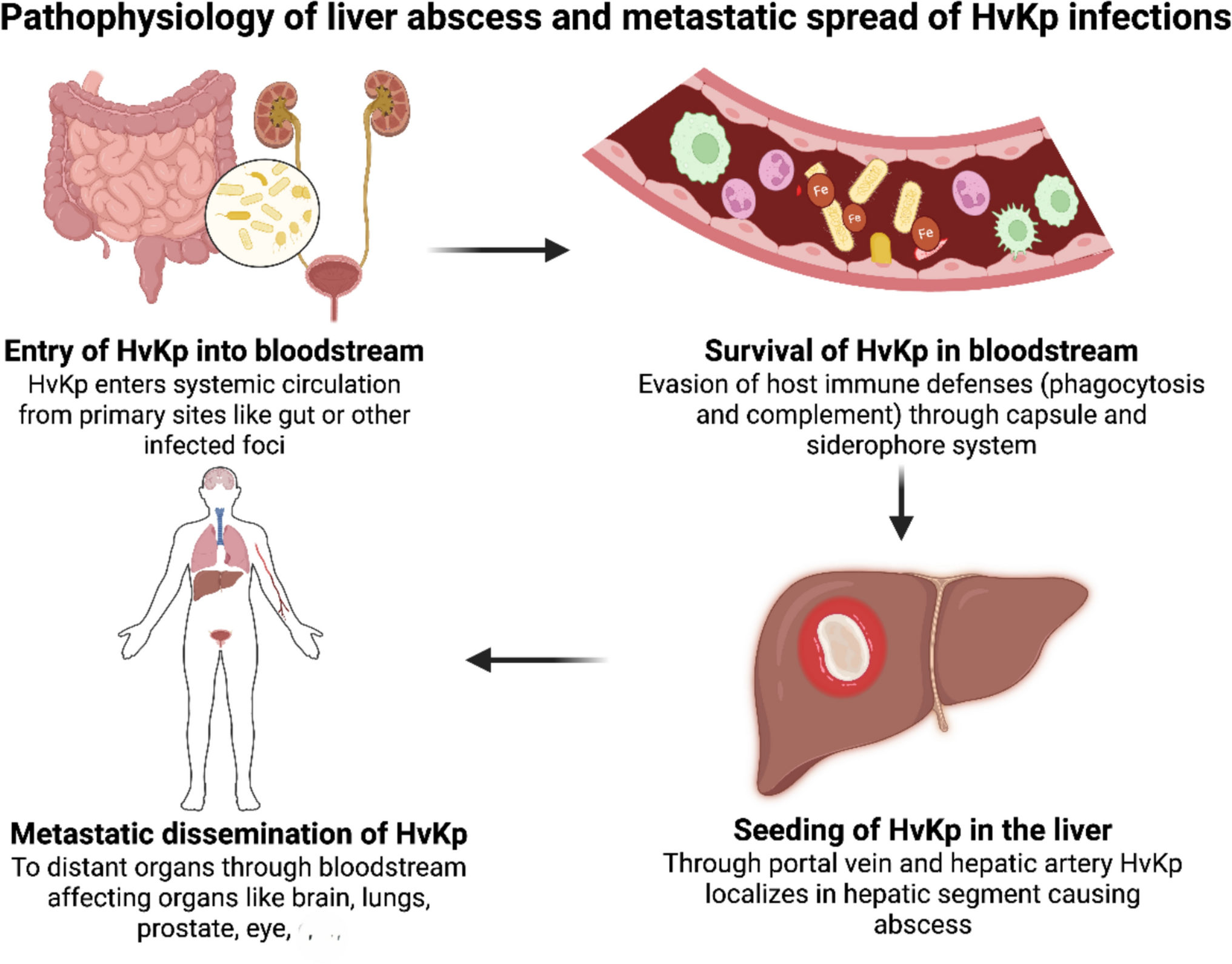
Figure 1 highlights the intricate connections between periodontal and systemic health, underscoring the critical need for early diagnosis, effective treatment, and a coordinated healthcare approach to enhance overall well-being of the patients.
Fig. 1
Original illustration depicting the pathways linking periodontal disease to systemic inflammation and associated health conditions. Periodontal pathogens, including Aggregatibacter actinomycetemcomitans (Aa), Porphyromonas gingivalis (Pg), Tannerella forsythia (Tf), Treponema denticola (Td), Eubacterium nodatum (En), Fusobacterium nucleatum (Fn), Prevotella intermedia (Pi), Campylobacter rectus (Cr), Peptostreptococcus (Micromonas) micros (Pm), Eikenella corrodens (Ec), Chlamydia pneumoniae (Cp), Prevotella nigrescens (Pn) and Capnocytophaga species (Cs), and their virulence factors, such as Lipopolysaccharides (LPS), disseminate from the infected oral cavity into the systemic circulation (pathogen dissemination). This process, along with the release of proinflammatory mediators including cytokines (Interleukin-1β [IL-1β], Interleukin-6 [IL-6], and Tumor Necrosis Factor-alpha [TNF-α]) and acute-phase proteins like C-reactive protein (CRP), contributes to systemic inflammation. This systemic inflammatory state is linked to various systemic diseases, including cardiovascular disease, diabetes mellitus, adverse pregnancy outcomes, and others discussed in this review. Immune responses involving cells like T-helper 1 (Th1) cells are also modulated
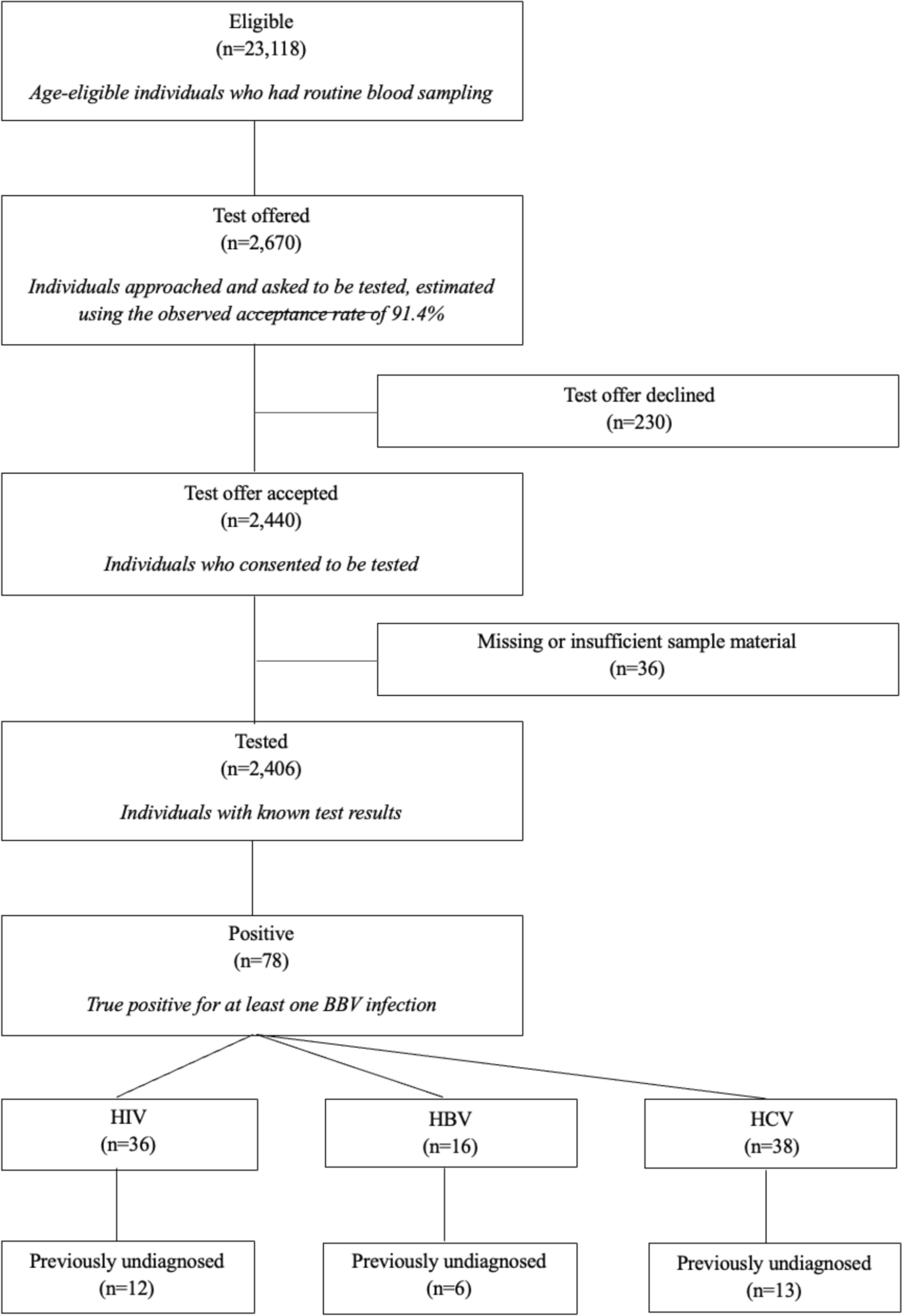
Cardiovascular diseaseCardiovascular diseases (CVDs), encompassing disorders of the heart and blood vessels, are the leading cause of death globally, accounting for an estimated 17.9 million deaths annually. Notably, there is a significant association between periodontal disease and an increased risk of cardiovascular events. Studies have shown that individuals with periodontal disease have a higher risk of experiencing heart attacks, strokes, or other serious cardiovascular events compared to those with healthy gums [62]. Pathogens such as Aa, Pg, Tf, Td, and Pi are transferred from the periodontal region to the vascular system (Fig. 2). These pathogens may also spread into circulation during normal oral activities or dental treatments that cause bacteremia. In circulation, these bacteria may adhere to the vessel walls and participate in the pathogenesis of atherosclerosis, resulting in plaque deposition in the vessel walls. This process is mediated by the inflammatory response that is stimulated by these pathogens [19, 20, 44].
Fig. 2
The diagram illustrates the association between periodontal pathogens, A. actinomycetemcomitans, P. gingivalis, T. forsythia, T. denticola, and P. intermedia and cardiovascular disease. These pathogens and their virulence factors translocate to vascular tissues, where they colonize and contribute to the formation of atherosclerotic plaques. Bacterial DNA from these pathogens has been detected in arterial plaques, linking periodontal infections to systemic vascular conditions and emphasizing the oral-systemic connection in disease etiology
A meta-analysis of five cohort studies involving 86,092 patients revealed that individuals with periodontal disease have a 12% higher risk of developing coronary heart disease (CHD). In contrast, case–control studies with 1,423 patients demonstrated an even greater risk, with an odds ratio of 2.22 [11]. An important piece of evidence supporting this relationship is the presence of DNA from pathogens such as Pg, Aa and periodontal Tf in atheromatous plaques, which proves that oral pathogens or their microbial products travel to other parts of the body [34, 35, 40]. Animal studies further corroborate this link, showing that oral infections with Pg and Td are associated with alveolar bone loss and aortic atherosclerosis, with bacterial DNA identified in systemic tissues and organs [63, 64]. Pg possesses specific virulence factors that are implicated in the development and progression of CVD. Its major cysteine proteases, known as gingipains, play a multifaceted role. Gingipains can degrade extracellular matrix proteins and components of cell–cell junctions, potentially compromising endothelial barrier integrity and contributing to endothelial dysfunction, a key early step in atherogenesis. Furthermore, gingipains can directly activate key elements of the coagulation cascade, such as prothrombin and Factor X, while also degrading fibrinogen. This dysregulation promotes a pro-thrombotic state, increasing the risk of clot formation (thrombosis) within atherosclerotic plaques [9, 58, 65]. P. gingivalis can also hide from the innate immune system, partly due to its unique LPS structure that weakly activates Toll-like receptor-4 (TLR-4), thus potentially maintaining chronic vascular inflammation rather than triggering acute clearance [40]. Additionally, P. gingivalis can induce platelet aggregation, contributing further to thrombosis risk. This occurs through both direct and indirect mechanisms: the bacterium's outer membrane proteins and vesicles can directly bind to platelet receptors (e.g., GPIb), triggering activation and aggregation, while inflammatory mediators released in response to the infection can also secondarily activate platelets [66, 67]. These mechanisms, particularly potent in Pg compared to some other oral pathogens like Aa or Td, underscore its significant contribution to the increased risk of atherothrombotic events in individuals with periodontal disease [65]. The presence of bacterial DNA within the plaques (Fig. 2) underscores the direct role of periodontal pathogens in the development and progression of atherosclerosis. Cytokines, such as interleukin-1β (IL-1β), tumor necrosis factor-alpha (TNF-α), and acute-phase proteins, including CRP, which are secreted during periodontitis, enhance systemic inflammation, which in turn aggravates endothelial dysfunction and plaque formation. This systemic inflammation not only worsens ather
Comments (0)