
Remember me
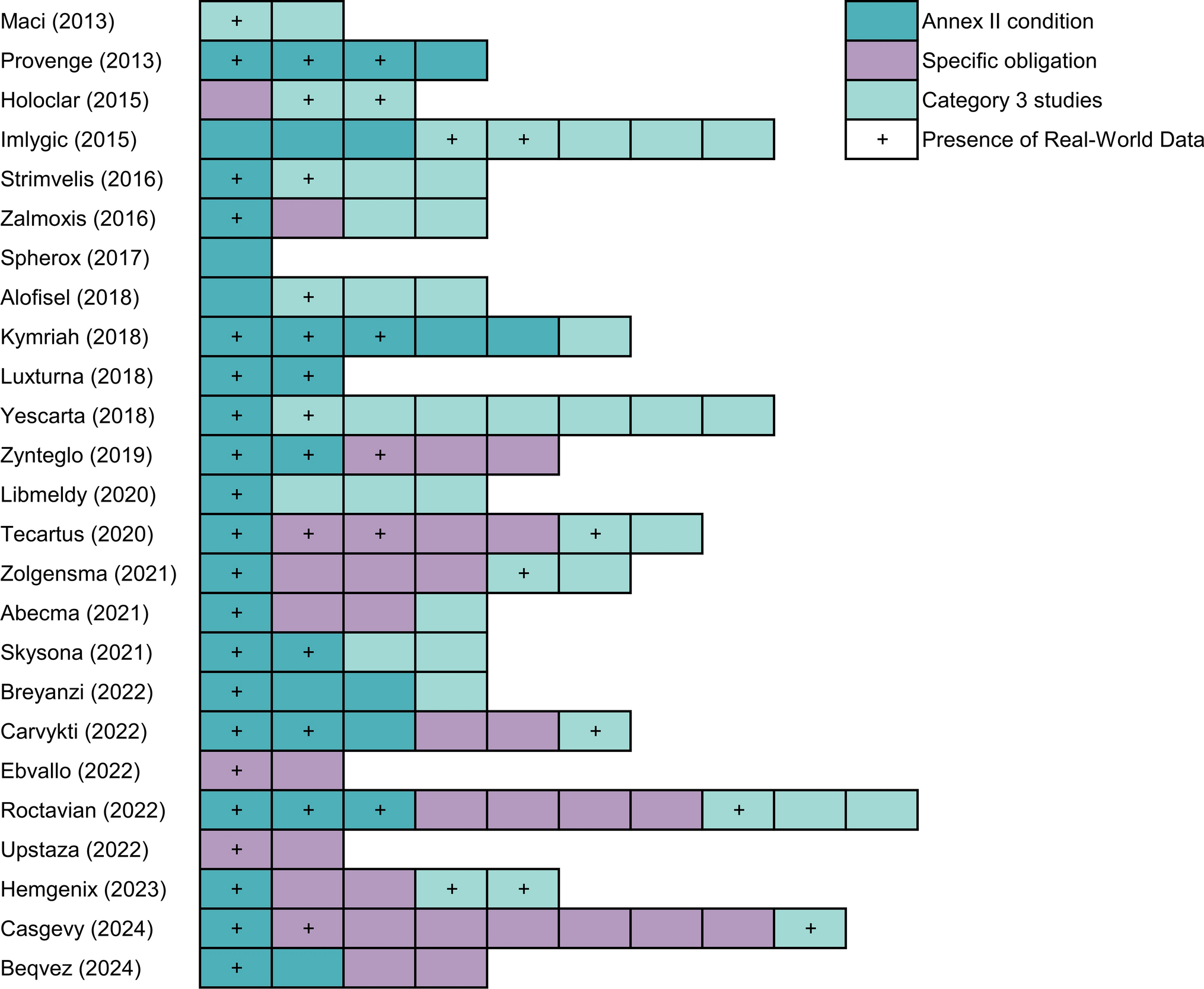


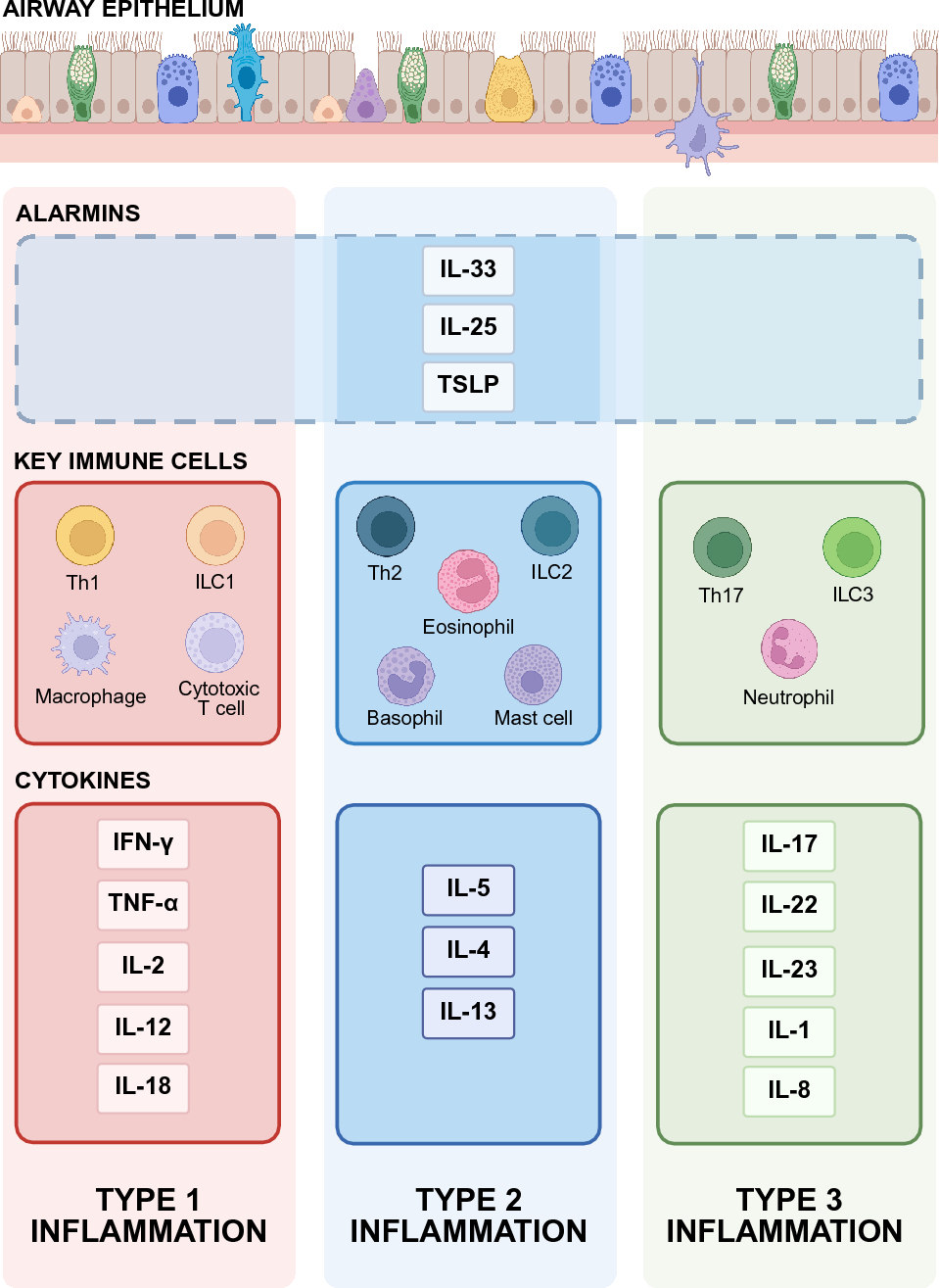


Across the four NGT discussions, a total of 21 participants attended. Table 1 presents an overview of participant characteristics.
Table 1 Demographic characteristics of the participants in this study3.2 RecommendationsIn total, 22 recommendations were developed, consisting of eight general (A–H) and 14 specific (1–14) recommendations. General recommendations reflect broader system-level priorities (e.g., stakeholder education, reliance mechanisms), while specific recommendations address analytical, nonclinical, and clinical parts of the biosimilarity exercise. Specific recommendations were categorized into six themes: (i) the reference product, (ii) analytical studies, (iii) nonclinical evaluation, (iv) clinical pharmacology studies, (v) comparative clinical efficacy studies, and (vi) local clinical studies. Of these 22 recommendations, 16 obtained high consensus, four moderate consensus, and two low consensus. Table 2 presents a summary of the recommendations, including weighted mean scores and consensus levels. Figure 2 highlights the highest-rated recommendations, showcasing key areas of strong stakeholder alignment. Detailed stakeholders’ perspectives and the underlying rationale for each recommendation is provided in Online Resource 2.
Table 2 Consensus recommendations with their weighted mean score from the Nominal Group Technique on recommendations for regulatory convergence and streamlined biosimilar developmentFig. 2
Schematic overview of the highest-consensus recommendations achieved among study participants for regulatory convergence and streamlining of biosimilar development
3.2.1 General RecommendationsThe recommendations are listed from highest to lowest obtained consensus.
A. Enhancing stakeholder education to build experience and confidence in the safety, efficacy, and quality of biosimilars.
Participants stressed that mutual understanding of science-based biosimilarity principles are essential to facilitate convergence. For regulators, targeted training on the rigor and robustness of comparative analytical and human PK data was considered critical to achieving alignment in regulatory decision-making and enabling further tailoring of clinical requirements. For healthcare professionals, particularly those involved in advisory committees, guideline development, or reimbursement decision-making, education was viewed as necessary to strengthen confidence in the scientific basis of regulatory approvals and to support acceptance and prescribing of biosimilars, particularly in the contexts where tailored clinical data requirements are applied. This recommendation received the highest consensus. While this recommendation does not directly address technical issues on streamlining biosimilar development, it emerged during the focus group discussions as a system-level challenge. Participants highlighted that the success of streamlined biosimilar development depends in part on broader stakeholder trust and shared understanding of science-driven biosimilarity exercise.
B–C. Promoting regulatory convergence and aligning requirements based on current scientific evidence.
Participants noted that regulatory convergence is essential to streamline biosimilar development and approval to create a consistent and predictable regulatory pathway. While agreeing that each national regulatory authority should retain autonomy, participants highlighted the importance of alignment on only science-based requirements.
D. Facilitating in-depth knowledge sharing among regulatory agencies.
Proactive, in-depth knowledge sharing, including case-specific findings from marketing authorization applications (e.g., if a biosimilar candidate demonstrates higher immunogenicity than its RP), was highlighted as crucial for supporting resource-limited regions. The International Pharmaceutical Regulators Programme (IPRP), and its biosimilar working group, were particularly recognized for facilitating collaboration and information exchange among regulators.
E. Integrating 2022 WHO biosimilar guidelines into national regulatory frameworks.
A multi-organizational approach was strongly advocated for regulatory convergence, emphasizing the complementary roles of WHO and ICH. Participants acknowledged that while the WHO is not a regulatory authority, its revised 2022 biosimilar guideline provides a globally accepted scientific framework that supports harmonization efforts.
F. Reliance on WHO-Listed Authorities operating at maturity level 4 for biosimilarity assessments.
Several participants noted that scientific alignment already exists across stringent regulatory agencies (e.g., EMA, FDA), and that the key barrier to convergence is often the absence of formal reliance mechanisms. The concept of reliance was identified as a strategic opportunity for regulatory convergence by enabling resource-limited regulatory authorities to leverage biosimilar assessments by other trusted authorities, especially WHO Listed Authorities operating at maturity level 4 (i.e., advanced level of performance and continuous improvement), without compromising national sovereignty [23].
G. Adopting a universally accepted template for public assessment reports.
Participants recommended developing a universally adopted template to enhance transparency and consistent documentation in public assessment reports for biosimilars by including standardized sections for different types of data, such as quality and clinical data. The existing IPRP template, known as the Public Assessment Summary Information for Biosimilars, was referenced as a useful example [24].
H. Developing International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use guidelines for biosimilar assessment.
Developing separate ICH guidance for biosimilar assessment was suggested to facilitate regulatory convergence. However, industry stakeholders raised concerns since biosimilar assessment is already based on ICH Q5E guidance, and this could undermine the principle of biosimilars being comparable to their RPs. Some participants also noted that ICH’s process, while extensive, may be too slow and rigid for the rapidly evolving biosimilar landscape.
3.2.2 Specific Recommendations3.2.2.1 The Reference ProductRecommendation 1: Harmonizing criteria for the selection of the reference product in biosimilar guidelines.
Participants noted challenges in procuring RPs for comparative testing, citing the high costs for clinical comparative efficacy studies that may not be scientifically justified, and practical barriers including limited market availability and originator companies’ reluctance to sell RP batches. A global comparator product was proposed to harmonize RP selection criteria. However, issues were highlighted about its implementation, including selecting the countries for sourcing and defining eligibility criteria.
Recommendation 2: Facilitating mutual recognition of reference products among WHO-Listed Authorities through data-sharing agreements.
Participants noted that despite some level of communication between regulatory authorities, developers may still need to obtain RP information themselves, such as manufacturing site details and specification variations. However, originator companies may be hesitant to disclose such details publicly. Therefore, participants recommended the establishment of data-sharing agreements, particularly between WHO-Listed Authorities operating at maturity level 4.
Recommendation 3: Establishing a unified, international consensus on the criteria for the type and number of bridging studies required in biosimilar development.
Many jurisdictions require analytical, and in some cases, clinical PK/PD bridging studies between locally sourced, foreign-sourced RP, and the biosimilar candidate. Participants highlighted inconsistencies in bridging studies requirements across regions and stressed the need for regulatory consensus on the type and number of required studies.
Recommendation 4: Establishing a stepwise approach to bridging studies in biosimilar development.
Since participants acknowledged that the cost of bridging studies is not trivial, duplicating these across multiple jurisdictions, adds substantially to the overall development costs. They suggested a stepwise approach, beginning with analytical bridging studies and advancing to PK bridging studies only if uncertainties are identified from the previous steps.
3.2.2.2 Analytical StudiesRecommendation 5: Implementing educational and training initiatives focusing on analytical studies.
Including compendial requirements for state-of-the-art and orthogonal analytical methods in biosimilar guidelines was discussed. While robust methods exist for evaluating critical quality attributes, participants agreed guidelines should focus on essential information. However, ensuring regulators have sufficient expertise to evaluate the analytical data package, was identified as a key challenge. Participants stressed the importance of education and training aligned with advancements in analytical methods.
Recommendation 6: Establishing “fit for purpose” criteria to ensure that the use of analytical methods contribute to meaningful support in biosimilarity demomynstration.
Participants discussed regulatory initiatives like the USFDA Research Roadmap for Biosimilars aiming to develop new analytical methods [25], and raised concerns regarding the overemphasis on increasingly sophisticated analytical methods. They suggested fit-for-purpose criteria to ensure that the choice of methods provide meaningful support for biosimilarity, rather than incremental improvements.
3.2.2.3 Nonclinical Evaluation: In Vivo Animal StudiesRecommendation 7: Establishing unified, international consensus regarding the non-requirement for in vivo animal studies
Some regions require in vivo animal studies, despite limited value and lack of scientific justification. Participants acknowledged the potential of validated animal models for specific purposes, such as assessing immunogenicity in complex biologics but stressed the need for regulatory consensus on minimizing in vivo animal studies to rare instances.
3.2.2.4 Clinical Pharmacology (PK/PD) in Healthy Volunteers or PatientsRecommendation 8: Establishing a scientific consensus on the validity of PD biomarkers.
Participants emphasized the early financial investments needed to identify and validate new PD biomarkers, along with challenges in analytical validation due to patient–patient variability and uncertainty in regulatory acceptability. They emphasized the need for regulatory consensus on the acceptability of new PD biomarkers.
Recommendation 9: Incentivizing the development and use of PD biomarkers.
Participants expressed concerns that regulators may consider PD biomarkers as a promising alternative to comparative clinical efficacy studies, emphasizing that expecting biosimilar developers, especially smaller companies, to invest in new PD biomarkers without success guarantee is rather optimistic. Consequently, the recommendation to incentivize PD biomarker development received the lowest consensus among all recommendations.
3.2.2.5 Comparative Clinical Efficacy Studies in PatientsRecommendation 10: Reconsidering the requirement of comparative clinical efficacy studies
Participants emphasized that comparative clinical efficacy studies often provide limited value in demonstrating biosimilarity, even for complex biologics such as mAbs and fusion proteins. They recommended scientifically justified criteria to determine their necessity and recognized exceptional cases where they may be warranted (e.g., when PK studies are not relevant, or additional therapeutic areas where more experience is needed). Furthermore, while the financial burden was noted as a contributing factor, particularly in cases where such studies are conducted despite limited scientific justification, this was framed within a broader concern regarding development inefficiencies that may impede development.
To reduce the reliance on comparative clinical efficacy studies when scientifically unjustified, participants suggested addressing the communication gap between varying disciplines among regulatory experts (e.g., those with quality and those with clinical expertise), who may lack a comprehensive understanding of the robustness of comparative analytical data. Implementing in-house training programs within regulatory agencies was recommended as a key strategy.
Recommendation 11: Harmonizing guidelines regarding clinical trial designs for biosimilars for the same reference product.
Participants highlighted varying regulatory requirements for clinical study designs and emphasized the need for standardized clinical trial designs for biosimilars of the same RP.
Recommendation 12: Relying more on the use of pharmacovigilance systems to monitor the safety and efficacy of biosimilars following market entry.
A few participants suggested relying more on pharmacovigilance systems to monitor biosimilar safety and efficacy in lieu of comparative clinical efficacy studies. This proposal received moderate consensus due to the importance of sufficient pre-approval evidence for biosimilarity and concerns about safety risks in regions with less robust pharmacovigilance systems.
3.2.2.6 Local Clinical StudiesRecommendations 13–14: Accepting clinical studies conducted for global submissions for biosimilar approval while evaluating the necessity of local clinical data based on observed ethnic differences with the reference product.
Participants noted that certain regulatory agencies, particularly in East Asia, require local clinical data for biosimilar approval. While ethnic sensitivity is typically addressed during the regulatory approval of the RP, participants recommended that local clinical studies should not be a default requirement for each biosimilar, emphasizing that the necessity for such data should be justified by observed ethnic differences with the RP. They acknowledged, however, that in jurisdictions where such requirements are legally mandated, legislative changes may be necessary to enable science-based regulatory decision-making.
Comments (0)