The purpose of this work is to inform pharmacoepidemiologists designing observational studies in China, South Korea, and Japan on the current regulatory landscape and to encourage continued discussion with regulators on the acceptance of alternatives to active surveillance studies such as hypothesis-testing studies. Routine pharmacovigilance forms a key part of the benefit-risk evaluation in China, Japan, and South Korea, where regulatory authorities have robust adverse drug reaction (ADR) reporting systems [3, 21, 28, 35]. The Korean Institute of Drug Safety and Risk Management (KIDS) was established in 2012. The KAERs system was developed by KIDS, which has stored all adverse events (AEs) since 2012. In addition to routine pharmacovigilance from ADR reporting systems, regulatory authorities in China, Japan, and South Korea generally request specific post-authorization studies (PAS) as a condition of the marketing authorization of pharmaceutical products. The submission of a risk management plan (RMP) summarizing the important identified risks, important potential risks, and missing information at the time of marketing authorization may inform the planning of PAS that can fill the knowledge gap about the benefit-risk profile of a product.
Globally, PAS is required by regulators to monitor AEs, fully characterize selected safety concerns, and confirm drug safety and effectiveness in patient populations that were excluded from clinical trials. In Asia, countries including China, Japan, and South Korea have established PAS requirements to confirm drug safety and effectiveness post-approval. These requirements are often due to concerns surrounding the small number of patients included in global registrational trials and possible genetic and environmental factors that may predispose certain populations to differential drug safety effects.
In Japan the re-examination procedure aims to reconfirm the safety and effectiveness of a pharmaceutical product in a defined time window. During the product filing, PAS may be mandated by the regulatory agency for implementation and completion during the re-examination period, with PAS results being submitted at pre-defined intervals during that period.
PAS may be designed as either primary data collection studies or as studies that make secondary use of data collected for purposes other than the study itself (e.g., database studies). Active surveillance studies (i.e., drug use result surveys, all-case surveillance) are usually observational single-arm descriptive studies requiring the systematic collection of all AEs and/or ADRs from the treated population in selected study sites. Additional data may also be required on a case-by-case basis to investigate the effectiveness or other aspects of the treatment regimen (such as hepatic or renal laboratory values). Patients are often enrolled in active surveillance studies through informed consent (though not required for regulatory-mandated PAS in Japan) and are treated with routine clinical care.
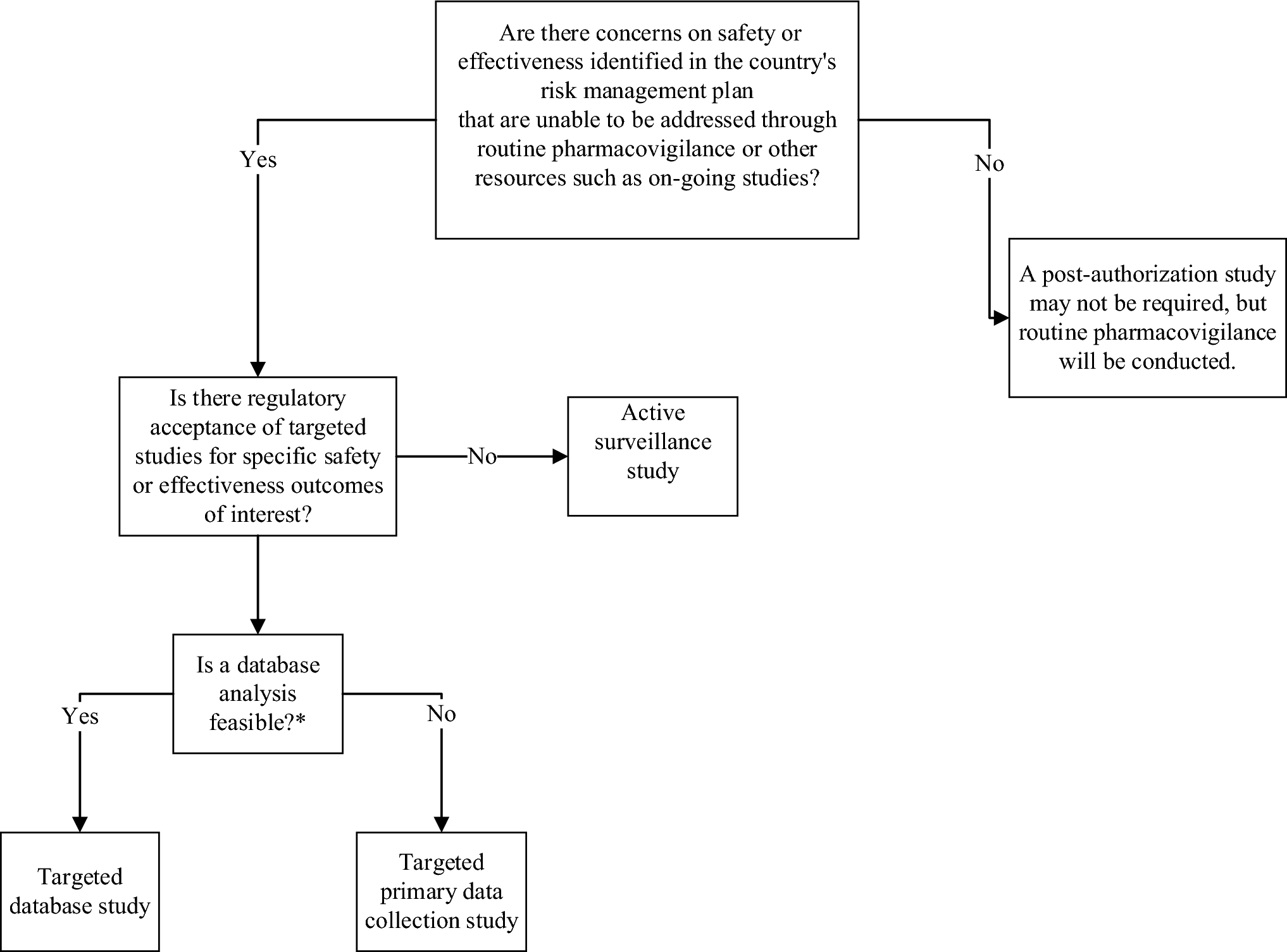
While active surveillance studies have been the traditional approach for post-authorization safety monitoring, there is a growing recognition of the value of conducting targeted studies that evaluate specific safety concerns identified in the RMP [11, 13]. Outcomes of interest for targeted studies may be identified via primary data collection or secondary use of databases. With the evolving diversity requirement in clinical trials and robust ADR pharmacovigilance systems, the question remains whether regulatory agencies favor targeted studies that evaluate specific safety concerns versus active surveillance studies that collect data on all AEs (ADRs).
Regulatory agencies in China, Japan, and South Korea have begun to embrace the conduct of targeted hypothesis-testing studies in databases compiling patient-level claims data or medical records. China, Japan, and South Korea have published regulatory guidance on using database studies to characterize the safety profiles of pharmaceutical products, indicating an increasing openness to considering alternative approaches to active surveillance studies.
Harmonizing regulatory approaches and enhancing access to comprehensive data sources are critical for generating fit-for-purpose evidence to support regulatory decision-making in these regions. Therefore, we propose a decision tool to assist with the planning of PAS in China, Japan, and South Korea. China, Japan, and South Korea were selected for this review based on author experience conducting PAS in these regions. Future publications reviewing PAS requirements in other countries in the Asia Pacific region would benefit the pharmacoepidemiology community.



Comments (0)