
Remember me


The IPNA Best Practice and Standards Committee has adopted the use of the RIGHT (Reporting Items for practice Guidelines in HealThcare) Statement for Practice Guidelines for the creation of CPRs [22]. A core leadership group (27 members including 18 pediatric nephrologists, 2 pediatric rheumatologists, 2 adult nephrologists, 1 kidney pathologist, 2 research trainees), an external expert group, and a voting panel were assembled. The core group conducted the systematic literature search and prepared the evidence review tables under the supervision of a pediatric nephrologist with expertise in epidemiology and two patient representatives. The expertise and responsibilities of the core group members are provided in Supplementary Table S1. The external expert panel consisted of 24 experts (22 pediatric nephrologists representing all IPNA regional societies, one pediatric rheumatologist, and one adult nephrologist). The core group provided a draft manuscript to the patient representatives, who reviewed it and provided feedback from relevant patient and family associations. The voting panel of representatives, selected from each IPNA Regional Society as experts in IgAN and IgAVN in children, was composed of 16 pediatric nephrologists and 2 pediatric rheumatologists (who voted on IgAVN only). Voting group representatives provided their level of agreement using electronic surveys which posted the question as per the Delphi method, and a 5-point scale with the options “strongly disagree,” “disagree,” “neither agree/disagree,” “agree,” and “strongly agree” was used. For topics that did not achieve a 70% level of consensus, the recommendations were then edited by the core group and voted on again until a consensus level of > 70% was achieved.
Developing clinical questions for the guidelineWe developed the questions to be answered in the guideline using the PICO format (Patient or Population, Intervention, Comparator, Outcome) and the following definitions: Population, children and youth (< 18 years) with IgAN or IgAVN; Intervention and Comparators, treatment compared with no treatment, other treatment, or placebo; Outcomes, changes in clinical biomarkers (urine protein, serum creatinine, estimated glomerular filtration rate (eGFR), kidney replacement therapy, kidney failure, interventions for treatment to induce remission or delay in progression of kidney disease).
Literature searchWe searched the PubMed database for relevant articles published by August 8, 2022. We retained all systematic reviews of randomized controlled trials (RCTs) on the treatment of IgAN and IgAVN in children, prospective uncontrolled trials, observational studies, biopsy classification and validation studies, and registry studies on the diagnosis and management of children with IgAN and IgAVN and restricted our search to human studies. Non-English language abstracts were also considered and translated into English whenever possible. Risk ratios (RR) with 95% confidence intervals (CI) were cited from two Cochrane systematic reviews evaluating RCTs of interventions for adult-onset IgAN and childhood IgAVN [23]. The literature search was also updated with relevant articles as they became available during manuscript preparation up to May 1, 2024. The publications used and a summary of the articles are provided in Supplementary Tables S2 and S3 for IgAN and Supplementary Tables S7 and S8 for IgAVN, while the search strategy is detailed in Supplementary Table S9.
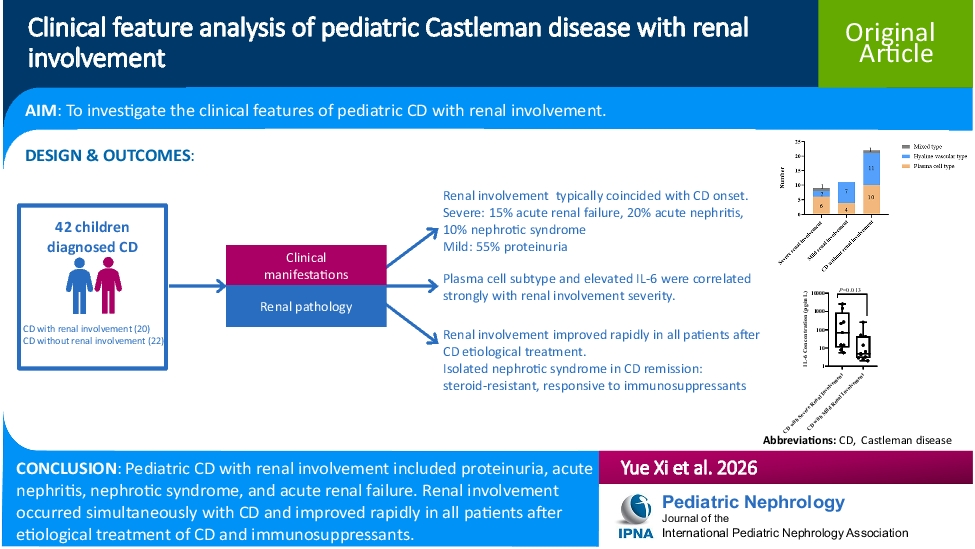
Grading systemThe American Academy of Pediatrics grading system was adopted by the IPNA Best Practice and Standards Committee [24] (Fig. 1). Each evidence statement was graded as high (A), moderate (B), low (C), very low (D), or not applicable (X) according to the system. Grade X refers to clinical situations where appropriate studies cannot be performed because benefit or harm clearly predominates; we also used it to grade clear contra-indications and safety boundaries. The strength of recommendations was also graded as strong, moderate, or weak.
Fig. 1
Matrix for grading of evidence and assigning strength of recommendations currently used by the American Academy of Pediatrics [22]
Clinical practice recommendations for IgA nephropathy (IgAN)Recommended definitions are provided in Table 1 and in the text as appropriate.
Table 1 Common definitions for IgA nephropathy (IgA) and IgA vasculitis nephritis (IgAVN)Initial assessment, diagnosis, and indications for kidney biopsyRecommendation 1a
We recommend obtaining a careful patient history for kidney and systemic manifestations and performing a detailed physical examination and blood and urine tests as per Table 2 in children suspected of having IgAN (grade A, strong recommendation).
Table 2 Initial clinical and laboratory workup and follow-up for children with IgA nephropathy.Evidence and rationaleIgAN can present with a range of clinical features, including asymptomatic microscopic hematuria (with varying degrees of proteinuria, with or without progressive kidney disease), intra-infectious (usually accompanying upper respiratory tract infection with fever, so-called “synpharyngitic”) macroscopic hematuria, rapidly progressive glomerulonephritis, nephrotic syndrome, and acute kidney injury (AKI) [4, 10]. IgAN is a condition that is characterized by the presence of dominant or co-dominant IgA staining in glomeruli. However, similar findings can also be observed in the context of other systemic illnesses, such as IgAV. These illnesses may present with symptoms such as rashes, oral ulcers, hemoptysis, abdominal pain, and joint swelling [2, 4]. KDIGO 2021 Clinical Practice Guidelines first recommend excluding IgAV and IgAN secondary to liver diseases, viral illnesses, and inflammatory bowel disease, as well as systemic autoimmune diseases and IgA-dominant infection-related (post-infectious) glomerulonephritis (GN) before diagnosing primary IgAN [2].
In patients with IgAN, decisions for management are mainly based on the amount of proteinuria, blood pressure levels, and eGFR. Urinalysis should document hematuria and quantify albuminuria and/or proteinuria (see Table 1 for definitions). For children, 24-h urine collection is difficult to perform. Thus, clinical practice recommendations from IPNA and KDIGO 2021 recommend assessment of (ideally) first-morning urinary protein/creatinine ratio (UPCR) instead of 24-h protein excretion in children with suspected glomerular diseases [2, 25]. An important differential in children presenting with proteinuria is orthostatic proteinuria which can be excluded through the collection of first-morning void urine samples to quantify proteinuria. First-morning void urine is recommended for all children with proteinuria to exclude orthostatic proteinuria. The eGFR should be calculated using the Schwartz formula and its modifications for children [26, 27].
Recommendation 1c
We recommend considering the possibility of primary IgAN, to be confirmed by kidney biopsy, in the presence of hematuria (gross and/or microscopic) with proteinuria (urinary protein/creatinine ratio (UPCR) ≥ 0.2 mg/mg or 20 mg/mmol) persisting over 2–3 weeks in at least two measurements on clear urine 1–2 weeks apart in the absence of lower urinary tract etiologies, or features of systemic disease, with a normal serum C3 level (grade X, strong recommendation).
Recommendation 1d
We suggest considering the possibility of primary IgAN, to be confirmed by kidney biopsy, in the case of persistent (> 2–3 weeks) or recurrent (> 2–3 times) gross hematuria occurring during an upper respiratory infection (as opposed to 2–3 weeks following the infection) (grade C, moderate recommendation).
Evidence and rationaleOften, the diagnosis of IgAN is suspected when children have abnormal urinary findings, especially in the school urinary screening program that is performed regularly in a few countries like Japan, South Korea, and Taiwan. An epidemiological survey based on urinary screening of children from Japan (n = 374,846) identified 37 children with IgAN with a mean age of 10.7 years [8]. Twenty-eight (75.7%) were biopsied due to an abnormal school urine screen while the remainder presented with gross hematuria; none presented with nephrotic syndrome or acute kidney injury (AKI) [8]. In a Japanese cohort of 258 children with IgAN, 62% presented with microscopic hematuria with or without asymptomatic proteinuria, 26% had macroscopic hematuria, and only 12% presented with acute nephritic syndrome or nephrotic syndrome [28]. On the other hand, in countries without a urinary screening program, most children with IgAN are identified after an episode of gross hematuria, most frequently concomitant with an infectious episode (classically an upper respiratory tract infection) [29]. In a study by the Southwest Pediatric Nephrology group from the USA (n = 218), 79% of children had gross hematuria at diagnosis, and 51% had proteinuria of 2 + or more on urine dipstick testing [29]. Also, a large series from Spain showed IgAN in 11.6% of 939 pediatric kidney biopsies, and macroscopic hematuria was the presenting symptom in 50.5% of these children [30].
Moreover, with the increasing availability of clinical trials and prospective registries for this condition, both requiring a histological diagnosis, a kidney biopsy is an absolute requirement for confirming a diagnosis in patients with suspected IgAN [2]. In patients with a positive family history of hematuria and CKD, or in patients with a diagnosis of “IgAN” with no response to adequate treatment, the possibility of collagen type IV mutations needs to be kept in mind, and performing genetic testing or α5(IV) chain staining on a skin/kidney biopsy specimen to explore this diagnosis is important [31,32,33].
Recommendation 1e
We recommend performing a kidney biopsy promptly in children with persistent (> 2–3 weeks) or recurrent hematuria and nephrotic-range proteinuria (UPCR > 2 mg/mg or 200 mg/mmol) and/or reduced eGFR (< 90 mL/min/1.73 m2) (grade X, strong recommendation).
We recommend performing a kidney biopsy in children with persistent (> 2–3 weeks) or recurrent hematuria and UPCR > 0.5 mg/mg (50 mg/mmol) in at least two measurements on clear urine 1–2 weeks apart (grade X, moderate recommendation).
We suggest performing a kidney biopsy in children with persistent (> 2–3 weeks) or recurrent hematuria and UPCR between 0.2 and 0.5 mg/mg (20–50 mg/mmol) in at least three measurements on clear urine 1–2 weeks apart (grade D, weak recommendation).
Recommendation 1f
We recommend considering the diseases listed in Table 3 as the major differential diagnoses of IgA nephropathy in children (grade X, strong recommendation).
Table 3 Major differential diagnosis of IgA nephropathy in children Explanatory note on kidney biopsyIn order to make a diagnosis of primary IgAN, a kidney biopsy that is processed for, at minimum, light microscopy and immunofluorescence (IF)/immunohistochemistry (IHC) for immunoglobulins (IgG, IgA, IgM) is required. Additional staining for C3, C1q, and C4 (all complement components); fibrin; and kappa and lambda light chains is recommended as per individual center practices. A diagnosis of primary IgAN requires dominant or co-dominant IgA glomerular staining among immunoglobulins, and the exclusion of differential diagnoses for clinical (IgAVN, secondary IgAN) and/or pathologic (e.g., IgA-dominant post-infectious GN) reasons (Supplementary Table S4). Electron microscopy is not required for diagnosis but is helpful in excluding hereditary basement membrane abnormalities and IgA-dominant post-infectious GN that is beyond the acute phase.
Evidence and rationaleAfter the exclusion of lower urinary tract etiologies (i.e., hypercalciuria, hypocitraturia, kidney stones, urinary tract infections), the main differential diagnoses to exclude in children with isolated, recurrent, or persistent hematuria are listed in Table 3. While some of these conditions can be distinguished from IgAN based on clinical and serologic findings, a kidney biopsy is needed to establish a definite diagnosis of IgAN and is often important for guiding therapy and assessing prognosis. A possible exception is X-linked Alport syndrome where a skin biopsy (with immunofluorescence studies for alpha-5 chains of type IV collagen) may often suffice, and all forms of Alport syndrome where a genetic study of COL4 mutations can provide a definitive diagnosis, especially if there is an appropriate family history [32, 33]. A recent history of potential post-infectious nephritis should also be sought. Figure 2 shows a flowchart for the diagnosis and management of IgAN for use in clinical practice.
Fig. 2
Management algorithm for IgA nephropathy
The use of immunosuppressive drugs, including glucocorticoids, before a kidney biopsy is not recommended unless a rapidly declining GFR suggesting rapidly progressive glomerulonephritis (see Recommendation 4b) is present, in which case it must be performed as soon as possible. The use of renin-angiotensin system blockers (RASB) to reduce proteinuria may be concomitant but should not delay performing a kidney biopsy. There is no single laboratory test or group of tests specific for IgAN. For assessment of kidney pathology, the KDIGO guidelines for adequacy of specimen and processing should be followed [2]. While the site of IgA deposits is best delineated by electron microscopy, a diagnosis of IgAN can be made based on light microscopy and IF/IHC findings alone.
Biopsies should be routinely classified according to the Oxford MEST-C classification (mesangial [M] and endocapillary [E] hypercellularity, segmental glomerulosclerosis [S], interstitial fibrosis/tubular atrophy [T], and crescents [C]). Classification categories are provided in Supplementary Table S5 [34]. While many studies have validated the use of this classification in adults, fewer studies in pediatric populations validate its use in this group with more variable results, likely related to smaller sample sizes and fewer patients reaching study endpoints (Supplementary Table S6) [35,36,37,38,39]. While mesangial, endocapillary, and extracapillary hypercellularity are common biopsy features in childhood IgAN, tubular atrophy/interstitial fibrosis, which is strongly associated with the development of kidney failure, is less frequent than in adults [40].
One study evaluating the causes of microscopic and gross hematuria in children from Thailand showed that among 342 children with microscopic hematuria, no cause was found in 276 children, and of the remaining 66, 16% had hypercalciuria while post-infectious (infection-related) GN was diagnosed in 1% [41]. Of those presenting with gross hematuria (n = 228), no cause was found in 86 patients while 22% had hypercalciuria and 15.7% were diagnosed with IgAN on biopsy [41]. In a retrospective review of 100 patients referred for gross hematuria (during 1992–1999), complete data was reported for 82 children; 13 (15.9%) were diagnosed with IgAN while 6 (7.3%) had thin basement membrane disease among those with glomerular hematuria (n = 24) [42]. A recent study identified 31% of patients with IgAN and thin basement membrane pathogenic variants in COL4A3/COL4A4/COL4A5 genes [43, 44]. Among those with non-glomerular hematuria (n = 56), 9 (16.1%) children had hypercalciuria [43]. A recent study observing 28 children with IgAN longitudinally supports the importance of kidney biopsy even in patients presenting with low levels of proteinuria [45].
Regarding children with recurrent macrohematuria and IgAN, evidence in children confirms the findings from adults that this clinical feature is accompanied by a better prognosis compared to patients with a single episode of isolated macrohematuria and to patients without hematuria [46]. However, persistent intense macrohematuria can occasionally, in IgAN patients as in other settings, be accompanied by AKI due to tubular necrosis. This is reported mainly in adult older patients, and a single pediatric report showed spontaneous resolution [47].
Clinical and laboratory workup, frequency of follow-upRecommendation 2a
We recommend long-term follow-up in children with IgAN, including those achieving complete remission, as they can relapse after many years (grade X, strong recommendation).
Recommendation 2b
We recommend adjusting follow-up intervals based on the severity of clinical symptoms, histopathological features, the treatment regimen, and the response to treatment (Table 2) (grade X, strong recommendation).
Recommendation 2c
We suggest re-evaluating the etiology of the disease to exclude secondary IgAN for children with persistent proteinuria (UPCR ≥ 0.2 mg/mg or 20 mg/mmol) after 3–6 months of supportive treatment (grade X, moderate recommendation).
Recommendation 2e
We recommend, upon diagnosis, offering the patient and his/her family psychological and social support, to be optimized on a case-by-case basis.
Evidence and rationaleThere is no robust evidence for the follow-up protocols of IgAN to improve kidney prognosis. Risk factors for disease progression include ongoing high levels of proteinuria, ongoing hematuria, hypertension during follow-up, decreased kidney function at the time of biopsy, and the histopathological grading [48]. Hypertension was a strong risk factor for IgAN progression. More severe histologic lesions are often found together with hypertension, higher amount of protein in the urine, and decreased eGFR at the end of follow-up.
Urinary findings can dynamically reflect the severity of disease in both adults and children with IgAN. Sustained proteinuria of 1 g/day was strongly associated with the progression of chronic kidney disease and with the development of kidney failure in IgAN in patients aged 15.6–73.5 years [48, 49]. Each gram per day increase above 1 is associated with a 10 to 25 times more rapid decrease in kidney function [49]. Remission of proteinuria might imply a reduced risk of progression to kidney failure, and proteinuria reduction early in treatment was associated with a lower risk for poor outcome, defined as the composite of time to the first occurrence of doubling of serum creatinine, kidney failure, or death [50, 51]. Based on more recent data from the RaDaR study, even lower levels of proteinuria (0.5–1 g/day in adults) appear to be associated with an increased risk of kidney disease progression [14]. In terms of hematuria, the lack of a validated and reproducible assessment modality has hindered studies on its prognostic value. Some single-center data suggest that remission of hematuria is associated with better kidney survival in IgAN [48, 52]. Microscopic hematuria may be an important risk factor for IgAN progression, as suggested by several studies recently reviewed and highlighted [53, 54]. However, recently, isolated recurrent macrohematuria has been shown to be a benign prognostic factor [46]. Regular urinalysis, documenting the presence and amount of proteinuria and hematuria, should be considered routine care for children with IgAN.
The amount of proteinuria, eGFR, blood pressure, pathological findings, and treatment regimen are used as a guideline for setting the regular follow-up interval. Close follow-up is required to detect eGFR decline among patients with IgAN at high risk of progression. Changes in kidney function should be monitored carefully when RASB are initially administered, and infectious risk should be monitored in patients receiving glucocorticoid or other immunosuppressive agents.
One study evaluating 96 pediatric IgAN patients with focal mesangial proliferation who were not treated showed that 57 (59.4%) achieved spontaneous remission of proteinuria and hematuria within a mean time of 5.9 years [55]. Of the 57 patients with spontaneous remission, ten (17.5%) recurred with urinary abnormalities. Maintenance of remission was observed in 79.9% and 67.9% at 5 and 10 years, respectively, after first remission. Severe clinical signs usually develop after 5 to 15 years [56]. Approximately 70% of patients who had childhood-onset IgAN had kidney symptoms even after approximately 20 years of follow-up evaluation [56]. A retrospective cohort study of 1012 patients (mean age of 32 ± 12.0 years) with IgAN at a single center in Japan showed that about 50% of patients progressed to kidney failure within 30 years despite treatment [57]. IgAN was falsely believed to be benign until one study in the 1990s reported that about 40% of IgAN patients progressed to kidney failure within 20 years [58, 59]. In a study of 241 Japanese children, it was reported that 5% of the patients had developed CKD by 5 years from the onset of the disease, 6% by 10 years, and 11% by 15 years [60]. Therefore, long-term follow-up is recommended in children with IgAN, even in cases with mild kidney dysfunction and mild proteinuria or in cases of complete remission, as urinary abnormalities may reappear and kidney failure may progress [2]. This has recently been confirmed by large-scale epidemiological studies evaluating adult risk of CKD in individuals presenting with different forms of kidney impairment, including glomerular diseases, in childhood [61, 62]. For this same reason, adequate psychological and social support is of paramount importance.
Some authors classify patients into low-risk groups and moderate-to-high-risk groups based on proteinuria, eGFR, and blood pressure, and suggest the follow-up interval should be every 6–12 months for at least 10 years for the low-risk group [63, 64]. The adult International IgAN Prediction Tool from the International IgA Nephropathy Network was recently updated for children, exploring an international cohort of 1060 children and concluding that this tool can also be used to assess the risk of progression in pediatric forms of IgAN [
Comments (0)