
Remember me



Ninety-nine patients with documented PXE disease are currently recruited in 2 French PXE expert centers. One is located at the university hospital of Nice (PI: Pr Georges Lefthériotis), and the other is located at the university hospital of Angers (co-PI: Pr Ludovic Martin). The centers were selected on the basis of their expertise and involvement in the management of PXE patients and the number of patients available for the study (n > 250 patients in Angers and Nice).
Sample size calculation follows an optimal Simon phase II design, including 2 randomized arms, with α = 10%, β = 5%, power = 95%, a 2:1 randomization ratio, to detect a success rate of 48% in the experimental arm. Clinical assumptions for the sample size calculation are based on literature data. Within the last decades, progresses in our knowledge of the PXE pathophysiology and recent studies conducted in the animals PXE models [23] and PXE patients [24] have brought results with the use of etidronate (a first-generation bisphosphonate).
At present, only one controlled/randomized trial has been performed (i.e., the TEMP trial: Treatment of Ectopic Mineralization in PXE) to evaluate etidronate (cyclical daily 20 mg/kg orally for 2 weeks every 12 weeks) versus placebo in 74 adults PXE patients (37 in each group) [24].
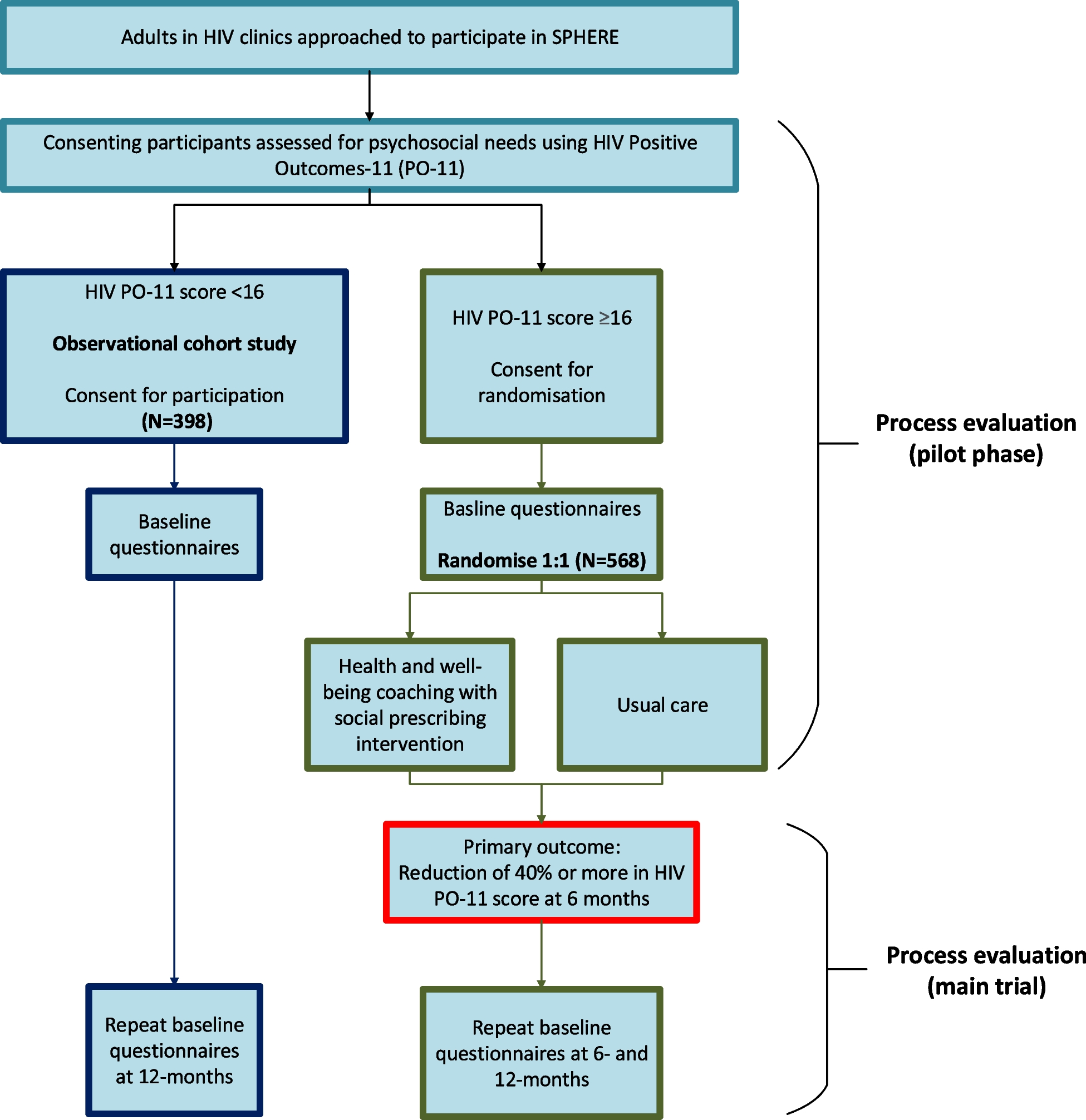
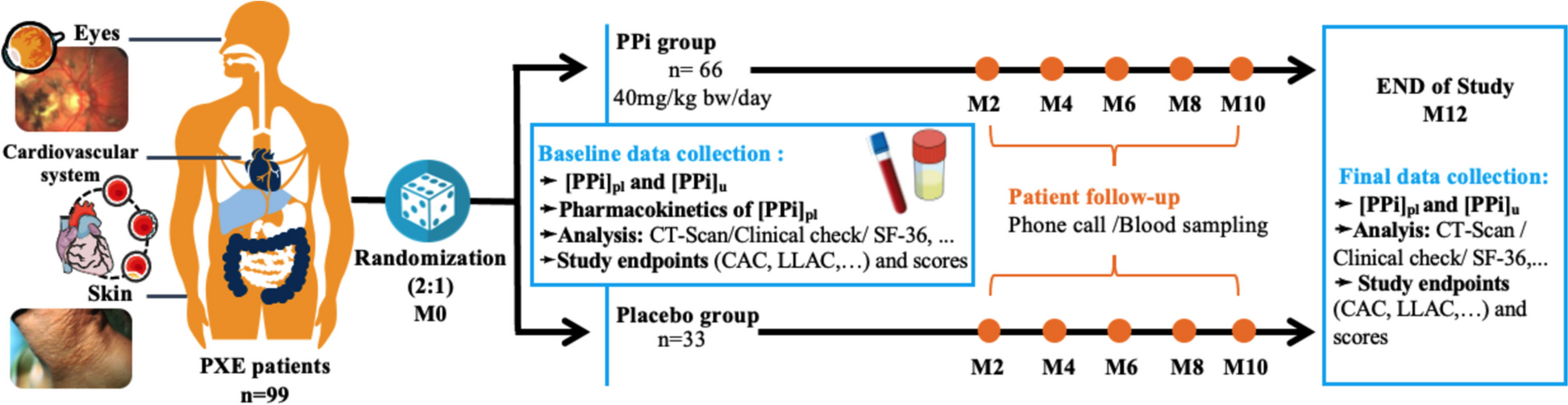
After screening for eligibility, signed informed consent (see additional file) is obtained from participants before entering the trial and randomly allocated to either treatment or placebo, (Fig. 1). This national bi-centric trial is recruiting patients since December 2022, its end being expected in 2026. This clinical trial protocol was designed following International Council Harmonization (ICH), Good Clinical Practices (GCP) guidelines and has been registered on the ClinicalTrials.gov website with the registration number NTC 04868578 (Fig. 2).
Fig. 2
PROPHECI clinical trial chart
Inclusion and exclusion criteriaPatients affiliated to the French social security scheme with a clinically confirmed and authenticated PXE according to the international diagnostic criteria [25], aged between 18 and 65 years, of both sexes, can participate in the study. Women of childbearing age must use an effective contraceptive method at inclusion and for the duration of their participation.
Patients are excluded on the following criteria (i.e., non-inclusion criteria): a positive serum pregnancy test at screening, lactating women or those who may wish to become pregnant during the inclusion period, patient < 18 years, patients suffering from osteomalacia, chronic diarrhea (> 1 month), impaired renal function (renal clearance < 30 ml/min/1.73 m2), electrolyte disorders (i.e., hyperphosphatemia and hypophosphatemia or hypercalcemia and hypocalcemia), vitamin D deficiency (< 35 nmol/L) or any other medical condition that may be considered incompatible in the opinion of the investigators, or enrolment in another clinical trial. Patients with major adverse effects of the trial drugs and who have withdrawn informed consent are excluded from the study (see additional file). During the trial, use of bisphosphonate, (e.g., etidronate) or other treatments for osteoporosis are prohibited.
Intervention drug and comparatorPatients are randomized in a 2:1 ratio. After randomization, patients receive double-blinded daily oral administration of encapsulated PPi salts (40 mg/kg bw/day of a 590 mg blend of food-grade Fe4(P2O7)3 and Na2H2P2O7 in gelatin capsules, ProNutri laboratory, Carros, France) or placebo (inactive ingredients) once in the morning, 30 min before or 2 h after a meal with a glass of water, and for 12 months. The daily dose of PPi supplementation was based on preliminary data from published absorption studies in mice and healthy human volunteers [20].
Criteria for discontinuing or modifying allocated interventionsNo adjustments to doses will be permitted throughout the trial for either treatment. Should a treatment interruption occur, it will be promptly documented in the logbook and communicated immediately to the investigators. Patients will remain enrolled in the study until its conclusion if a treatment interruption becomes necessary.
Strategies to improve adherence to interventionsTo ensure adherence, patients will receive bi-monthly phone calls as part of the clinical trial’s compliance monitoring protocol. Evaluation of compliance will rely on weekly logbook entries completed by the patients, detailing adherence, adverse events, and other relevant information.
Provisions for post-trial careFollowing the trial, patients will resume their standard disease monitoring. As per French law, no direct financial compensation is provided for the trial. However, compensation is available in case of harm through the sponsor’s insurance.
Study outcomesPrimary outcome measuresThe primary objective was to evaluate the progression/regression of the calcification process using 18F-NaF PET. Based on these previous data, we were able to calculate the sample size. The efficacy of PPi supplementation is gauged by alterations in calcification scores derived from non-contrast CT scan imaging between baseline (M0) and after 12 months of treatment (M12). The percentage of PPi and placebo-treated patients experiencing regression or stabilization of their calcification scores, compared to those experiencing progression, will be analyzed following an optimal Simon phase II design [26].
Secondary outcome measuresEfficacy on peripheral arterial disease (PAD) is assessed on the Welch auto-questionnaire [27], the ankle-brachial pressure index [28], and treadmill walking distance [29]. Efficacy on progression of ocular lesions is based on the worsening of visual symptoms and/or the number of anti-VEGF ocular injections performed at baseline and after 12 months of treatment. Skin lesions are assessed by a clinical scoring by trained dermatologists, with a dermoscopic analysis. Treatment safety is evaluated according to the Common Terminology Criteria for Adverse Events (CTCAE) v4.0. In addition, quality of life is evaluated on changes in the SF-36 questionnaire score [30].
Randomization and blindingInclusion and randomization are not performed at the same time. The investigator or its delegate enrolls and randomizes patients using e-CRF (see additional file). Randomization is stratified by a centralized blocked balanced randomization according to a 2:1 ratio, managed by the Clinical Research Unit (URC-EST). To maintain blinding, patients, investigators, study staff, and biostatisticians are unaware of treatment allocation. Both experimental and placebo capsules are placed in identical-looking bottles labelled with trial information (promotor name, protocol name and number), treatment name, and number, and an empty patient number, which will be filled in by study nurses. Patients will be instructed to take the predefined number of capsules of their assigned medication daily.
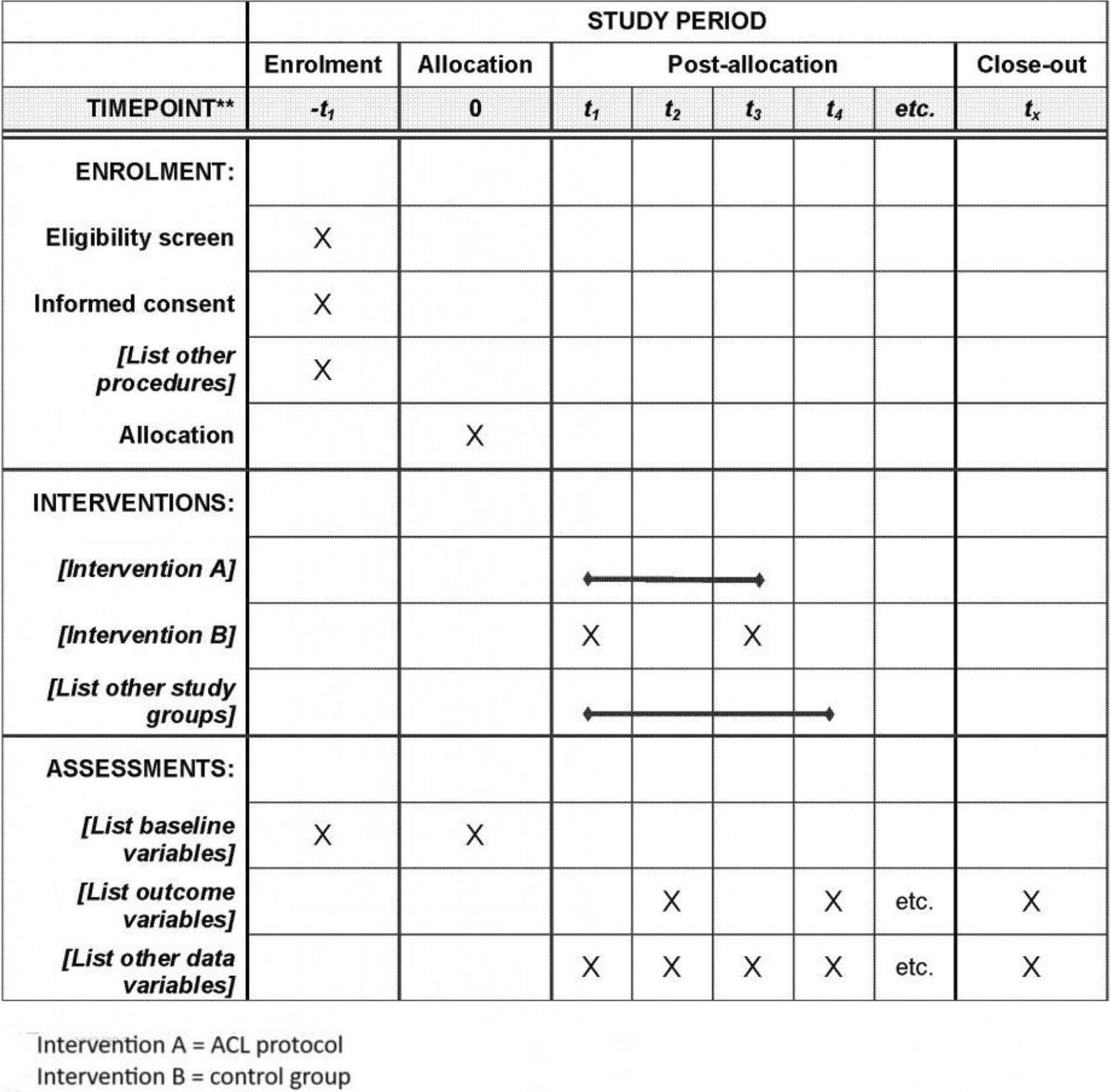
Participant timelineOn the day of enrollment, all participants undergo complete physical and specialized examinations, non-contrast CT scan, blood, and urine analyzes (calcium, phosphate, magnesium, chloride, blood osteogenic activity: PTH, osteocalcin, alkaline phosphatase (ALP), vitamin D (25 OH D3 and 1.25 OH D3)). Plasma PPi concentration is specifically quantified using our laboratory’s established method [8, 31]. Throughout the study period, all medical and surgical events, including changes in medications, and adverse events are documented via phone calls. Adverse events are scored according to CTCAE v4.0. Adverse effects will be evaluated along the study from the first day of administration to the latest.
The participant timeline is outlined in Table 1, adhering to the SPIRIT reporting guidelines [32].
Table 1 PROPHECI study flow chart from enrolment (M0) to end of study (M12)Data collectionIn compliance with the standard operating procedure in place in our data management center (Centre Antoine LACASSAGNE, Nice, France), the case report form for this clinical investigation was designed after the protocol had received the approval of all persons who had participated in drafting the protocol. The participation of our DEBDS (Department of Epidemiology Biostatistics and Health data) and data management center (Centre Antoine Lacassagne) was requiered to:
Create the electronic case report form, which will be considered as the Database, using the Ennov Clinical software developed by the ENNOV group (https://fr.ennov.com/)
Ensure quality control and export of patient’s data
The Ennov Clinical software ensures traceability of access and data modifications, thanks to an audit trail. Each access is ensured by a unique identifier and password for each user, who has specific rights according to his/her role.
The Ennov Clinical software meets the strict requirements defined by the Food and Drug Administration (FDA) guidelines in chapter 21, CRF, Part 11.
The database will be hosted by ENNOV data centers on secured servers (two backup sites located on different geographical sites). Before being processed by the biostatistician commissioned by the sponsor, the data manager will check the data using consistency tests (queries) and ensure that any inconsistencies detected are resolved. All data will be anonymized and treated as strictly confidential.
Biological samplePatients participating will provide blood samples from a peripheral vein (8 tubes, 16 mL samples) and urine (10 mL sample) at the baseline (M0) and after 12 months of treatment (M12). These samples will be collected for standard blood/urine analysis and PPi or other biological determinations. Blood tubes for PPi determination will be promptly transferred on ice for specific pre-analytic treatment by Anger and Nice PXE centers according to the LP2M laboratory’s protocol (WO2023036949A1). Ultra-filtrated plasma samples will be frozen – 80 °C for biological repository and transferred to LP2M laboratory, where analysis will be conducted in a blinded manner.
Quantification of arterial and renal calcificationCalcification score from non-contrast whole-body CT scan will be performed using the Agatston score method [33] with a threshold of 130 Hounsfield Units. Calcifications in the peripheral arterial segments including carotids, ulnar and radial arteries, coronary arteries, aorta, femoropopliteal arteries, and below-knee arteries are quantified and normalized to the arterial wall surface (data expressed as HU/mm2 of arterial surface). The interclass correlation coefficient for inter-observer reliability of the total peripheral artery calcium mass measurement is determined by a scoring of 10 random scans by 2 independent investigators. Inter-observer reliability and inter-class correlation coefficients for the total peripheral artery calcium mass measurement is determined by 2 independent investigators scoring 10 random scans. Additionally, quantification of renal calcifications will be performed on the same CT scans by expert [12, 21].
Vascular studiesAnkle-Brachial Index (ABI) will be determined at rest by the ratio between the lowest ankle artery pressure and the highest brachial artery blood pressure [28]. A peripheral arterial occlusive disease (PAOD) is defined according to the actual recommendations [34]. Absolute Walking distance will be assessed on a treadmill and completion of a Welch auto-questionnaire [27].
Ophthalmologic studyChanges in ophthalmologic parameters are assessed by the occurrence of subretinal neovascularization events, the frequency of anti-VEGF administration, and best corrected visual acuity (BCVA). A subretinal neovascularization event will be defined by the need to initiate or intensify anti-VEGF injections to prevent (further) visual impairment, i.e., retinal bleeding suspected to be caused by subretinal neovascularization, if necessary, confirmed by fluorescein angiography, a significant increase in subretinal or intraretinal fluid and growth of a subretinal neovascular complex. These events will be evaluated by trained ophthalmologist at the PXE reference centers who will be blinded to treatment status.
Dermoscopic studyDermatological changes will be objectified by a dermoscopic study of the skin according to the following protocol. Images will be acquired from commonly affected areas (neck, axilla, antecubital fossae, armpits, and periumbilical area) by a contact dermoscopy (DS) with polarized light and recorded, as previously described [35]. The investigators will ensure that the very same areas are imaged at baseline and after the treatment are completed. The surface of DS elementary changes featuring yellow papulae (presenting as “dots” or “reticular networks”) on digitized images will be compared using an open image processing software (ImageJ, https://imagej.nih.gov/ij/index.html NIH).
Life quality assessmentTransformed SF-36 scores between 0 and 100 on the 9 quality-of-life domains will be used: physical function, physical role functioning, pain, general health, mental health, social function, social role functioning, vitality, and health changes [30].
Statistical analysisThe main objectives of the study will be analyzed after inclusion of the 62 patients. The success/failure rate in the experimental arm will be calculated (see additional file). Success will be defined as patients experiencing regression or stabilization of their calcification score between the first and twelfth month post treatment. Failure will be defined as patients experiencing progression of their calcification score over the same period. According to the optimal Simon phase II design [26], if there are 29 or fewer patients experiencing success, the assumption of PPi efficacy will be rejected. If there are 30 or more patients experiencing success, PPi efficacy will be confirmed. An interim analysis is planned after the inclusion of 27 treated patients (Fig. 3). Considering that 5% of the patients will be lost to follow-up, 62 patients were required to detect a success rate of 73% in the experimental arm. Clinical factors influencing success/failure rate will be tested via univariate analysis. Multivariate analysis will be performed if needed. All statistical analyzes of secondary objectives, i.e., the safety of PPi in PXE, on ophthalmic lesions, and the efficacy of PPi on peripheral arterial disease and on skin lesions will be evaluated according to CTCAE v4.0.
Fig. 3
Simon’s interim analysis
Comments (0)