
Remember me

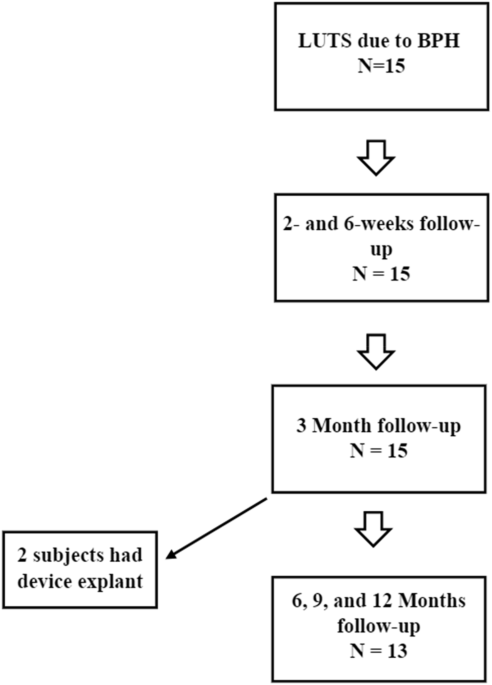
This was a single-center, single-arm study to evaluate the technical feasibility and safety profile of the FloStent™ System in men ≥45 years of age with BPH. Descriptive statistics were employed to examine perioperative data, acute safety, and rapidity of symptom relief. The primary endpoints of the study were device implantability, device tolerability, and device retrievability. The original protocol called for a three-month study duration, with all subjects originally consenting to planned removal of the stent at the three-month timepoint. However, due to encouraging safety and efficacy data collected at the initial follow-up visits, the original protocol was amended to allow subjects to retain the stent for up to one year. All subjects were thus given the option to undergo device removal at three months, per the original protocol, or continue in the study and undergo device removal at 12 months, per the amended protocol (Fig. 1). Those subjects opting for study extension provided a separate informed consent for the study extension. Because the study included first-in-human device use, and all subjects were potential candidates for BPH intervention, all subjects were offered surgical standard-of-care therapy (TURP) after device explant, in order to ensure standard-of-care treatment was ultimately delivered. The protocol and all amendments (Rev 1.0, 1.1, 2.0, 2.1) were approved by local institutional review board (Comite de Bioetica de la Investigacion del Instituto Commemorativo Gorgas, CBI-ICGES) and national health ministry in Panama.
Fig. 1
The study cohort was enrolled as a first-in-human (FIH) trial. The primary focus was implantability and safety. As such, participants were recruited based on either IPSS of >15 or peak urinary flow rate (Qmax) of ≤12 mL/s.
The formal inclusion criteria for this study encompassed men ≥40 years of age, with a prostate volume range of 25–80 cc, an International Prostate Symptom Score (IPSS) greater than 15 or a peak urinary flow rate (Qmax) of 12 mL/s or less, and a prostatic urethral length range of 2–4.5 cm (visualized via cystoscopy). Exclusion criteria included prostatic urethral length less than 2 cm, estimated prostatic volume less than 25cc or greater than 80cc, presence of an obstructing intravesical prostatic median lobe, urinary incontinence due to an incompetent external sphincter, and urethral pathologies preventing insertion of the delivery system, i.e. urethral stricture. Additional exclusion criteria included current symptomatic urinary tract infection or significant visible hematuria and subjects with a known allergy to nickel or titanium. Participants were also excluded if they had a known or suspected urological condition other than benign prostatic hyperplasia affecting voiding function, or neurogenic bladder and/or sphincter abnormalities.
Follow up assessments were performed at visits at 2, 6, 12, 26, 36, and 52 weeks. Though the study was not designed to make efficacy claims or test a hypothesis, clinical parameters including IPSS, Qmax, PVR, International Index of Erectile Function (IIEF), and Visual Analog Scale (VAS) were collected at baseline and at each follow up assessment. Additionally, the VAS was collected at the time of procedure.
FloStent implantationThe patient was placed in supine position and a transabdominal or transrectal ultrasound was conducted to confirm prostatic size. Perioperative antibiotics based on local antibiotogram and intravenous and/or local anesthesia (intra-urethral gel) were administered. A diagnostic flexible cystoscopy was performed to assess prostatic urethral anatomy. Prostatic urethral measurement was conducted using the cystoscopic pullback technique, measuring from the bladder neck to verumontanum, after which the cystoscope was withdrawn. Implant size was selected based on the measured prostatic anatomy. Once the cystoscope was within the prostatic urethra, the delivery tool was used to push the stent out of the cystoscope’s working channel. Once the stent was fully exposed and within proper position, the actuator on the grasper or delivery tool was used to release the implant. The delivery tool was then withdrawn. The cystoscope was used to assess implant patency and position. The stent when properly positioned was placed fully within the prostatic urethra, without crossing the bladder neck or external urinary sphincter (Fig. 2). If needed, stent position was adjusted using standard grasping forceps or by utilizing the tip of the flexible cystoscope. Once the implant position was visually satisfactory, the cystoscope was withdrawn. Voiding was confirmed via post void residual prior to postoperative discharge.
Fig. 2
Endoscopic view of implanted FloStent.
FloStent retrievalDevice retrieval (explant) was safely performed in all patients. Two devices were removed per the original protocol at 12 weeks. Both patients declined TURP and were doing well at time of last clinical follow-up. 13 devices were removed at 52 weeks per the amended protocol (study extension). Twelve patients underwent TURP at the time of device retrieval, which was performed with a bipolar electrosurgical loop. In the remaining patient, TURP was deferred by the investigator due to a narrow penile urethra which would have required calibration with sounds; the patient reported good voiding after device removal and thus far has not pursued TURP.
Since this study represented the first experience with device retrieval in humans, retrievals were performed under general anesthesia in all patients. Two devices (both at 52 weeks) were removed with a flexible cystoscope; the remainder were removed with a rigid cystoscope. Standard graspers were used in all cases. No electrosurgical or laser energy was required for device retrieval. Minimal to mild mucosal bleeding was observed immediately post-explant.
In patients who did not undergo TURP at time of device explant, light cautery was used to control bleeding at the explant site. A postoperative catheter was left in place overnight in these patients.
Comments (0)