
Remember me
This research complies with all relevant ethical regulations as detailed in the relevant sections below.
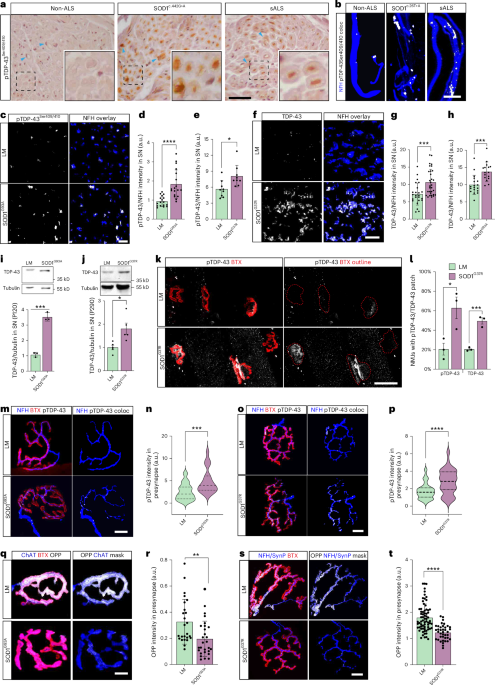
Human muscle biopsy for intramuscular nerve staining and human obturator nerveIntramuscular nerve staining was performed on muscle biopsies from a patient with SOD1 ALS, a patient with sALS and a male non-ALS patient (Supplementary Table 5). Obturator nerve biopsies were obtained from a patient with SOD1 ALS, a patient with sALS and a non-ALS patient (Supplementary Table 5). The local ethics committee of the San Raffaele Hospital on human experimentation approved the study protocol (RF-2019-12369320); obturator nerves were collected and stored in our tissue bank, after informed consent, both for patients with ALS and for controls. All clinical and muscle biopsy materials used in this study were obtained with written informed consent during 2016–2020 for diagnostic purposes followed by research application, approved by the institutional review board. Biceps skeletal muscle samples were excised via open biopsies, and pathological analysis was performed at the Hacettepe University School of Medicine, Neuromuscular Diseases Research Laboratory, in Ankara, Turkey (Hacettepe University ethics committee approval no. GO20/177, 11/02/2020) and at the neuromuscular pathology laboratory at Sheba Medical Center in Ramat-Gan, Israel. All procedures involving human participants were approved by the Sheba Medical Center institutional review board, with approval numbers 0004871-18, 0001680-7 and 0001681-7, in accordance with the Declaration of Helsinki. Patients with ALS and non-ALS patients were all Caucasian males, aged 26–66 years (48.33 ± 13.12). Patients with ALS were diagnosed with clinically definite or probable ALS according to El Escorial criteria70. Non-ALS control muscles included a variation of findings, which were consistent with a diagnosis of normal muscle, severe, chronic ongoing denervation and reinnervation due to spinal stenosis.
Frozen muscle biopsies were cryosectioned to 10-µm-thick slices, mounted onto slides and air dried for 30 minutes at room temperature. Sections were washed in PBS, fixed in 4% paraformaldehyde (PFA) for 20 minutes, permeabilized with 0.1% Triton and blocked with 5% goat serum (The Jackson Laboratory) and 1 mg ml−1 BSA (Amresco). Sections were then incubated with appropriate antibodies overnight at 4 °C in blocking solution: rabbit anti-phosphorylated TDP-43 (1:1,000; Proteintech) and chicken anti-NFH (1:1,000; Abcam). Sections were washed again and incubated for 2 hours with secondary antibodies (1:1,000; The Jackson Laboratory and Thermo Fisher Scientific), washed and mounted with ProLong Gold (Life Technologies).
For obturator nerve biopsies, the anterior motor branch of the obturator nerve was sampled and processed in all patients according to our standardized protocols15,71
Experimental animalsSOD1G93A−ChAT::tdTomato and LMChAT::tdTomato embryos were obtained by mating ChAT::ROSAtdTomato females with SOD1G93A males. Embryos were genotyped to distinguish wild-type from SOD1G93A embryos. The colony was maintained by breeding males and females within the colony. HB9−GFP (The Jackson Laboratory, stock no. 005029) mice were originally obtained from The Jackson Laboratory. The colony was maintained by breeding with ICR mice (Institute of Animal Science, Harlan). B6SJL.SOD1G93A (The Jackson Laboratory, stock no, 002726) colonies were maintained by breeding with C57BL/6J mice. Genotyping was performed using PCR (Kapa Biosystems), and DNA samples were generated from ear or tail tissue biopsies. B6SJK.SOD1G93A and littermate males were used to characterize TDP-43 pathology. For the in vivo miR-126 and GFP PHP-eB AAV injections in B6SJK.SOD1G93A and littermates, four female and three male mice of B6SJK.SOD1G93A and five female and three male littermate mice were used. All animal experiments were approved and supervised by the Animal Ethics Committee of Tel Aviv University. Colonies of LoxSOD1G37R mice (The Jackson Laboratory, stock no. 016149) harboring a floxed transgene comprising human SOD1 gene bearing G37R mutation72,73 were maintained by breeding heterozygous loxSOD1G37R males with females with pure C57BL/6J background to eliminate confounding genetic influences. LoxSOD1G37R and littermates of both sexes were used to characterize TDP-43 pathology. B6.SOD1G93A (The Jackson Laboratory, stock no. 004435) colonies were maintained by breeding with female C57BL/6J mice. Mice were genotyped by PCR of DNA extracted from a tail biopsy. B6.SOD1G93A and littermate males were used to characterize TDP-43 pathology. All animal procedures were approved by the Institutional Animal Care and Use Committee of Ben-Gurion University of the Negev, in compliance with Israel’s Animal Welfare Act (1994) and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (National Research Council, 2011). The animal facility is approved by the US Office of Laboratory Animal Welfare (assurance no. A5060-01) and is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Differentiation of iPSCs into functional MNs, myotubes and neuromuscular co-cultureSOD1A5V and TDP-43M337V iPSC lines used in this study were obtained from Michael Ward (NIH) and the iPSC Neurodegenerative Disease Initiative. The use of these lines was approved by the institutional ethics committee of Tel Aviv University (approval no. 0001160). All lines were derived with informed donor consent under NIH protocols and in accordance with the Declaration of Helsinki. iPSC colony maintenance and differentiation into MNs was performed as previously described13,74. In brief, MN transcription factor cassette including the transcription factors islet 1 (ISL1) and LIM homeobox 3 (LHX3) along with NGN2 were integrated into a safe harbor locus in iPSCs under a doxycycline-inducible promoter74.
Skeletal myotube differentiation was performed as follows. Direct myogenic conversion of human iPSCs was performed by transfection of iPSCs using a doxycycline-inducible MYOD1-shOct4 cassette as described, and this cassette was a gift from Michael Ward (Addgene plasmid no. 182309; http://n2t.net/addgene:182309; RRID: Addgene_182309). iPSC clones were cultured in six-well plates coated with Matrigel (Corning, 356234), grown in mTesr1 medium (STEMCELL Technologies, 85850) and passaged with mTesr1 medium containing 10 μM Rho-kinase inhibitor (RI) (Sigma-Aldrich, Y0503) for 1 day after passaging. Culture media were refreshed daily until colonies reached 80% confluence. For differentiation, 300,000 iPSCs were plated in a 35-mm dish coated with Matrigel (1:100) in mTesr1-RI medium. On the following day, media were replaced with induction medium supplement containing DMEM/F12 (Gibco, 31330038), 1× sodium pyruvate (Gibco, 11360070), 1% NEAA (Biological Industries, 01-340-1B) with 10 μM RI, 0.1 mM β-mercaptoethanol, 2 μg ml−1 doxycycline (Sigma-Aldrich, D9891) and 10 μg ml−1 recombinant human insulin (R&D Systems, 11376497001). This induction medium is replaced every day for two consecutive days, and then half-media volume is replaced for every alternate day until day 6. After the first 2 days of differentiation, RI is removed from the induction medium. On day 7, fresh induction medium containing 3 μM CHIR99021 is added for 48 hours along with induction medium. Followed by CHIR treatment, induction medium is replaced by maturation media containing the following: Neurobasal A medium (Gibco, 10888022), 1× B27 Plus supplement (Gibco, A3582801), 1% NEAA (Biological Industries, 01-340-1B), 1% Optimal-Culture-One supplement (Gibco, A3320201), 0.1 µg ml−1 Agrin (R&D Systems, 6624-AG-050), 0.05 µg ml−1 Sonic Hedgehog (PeproTech, 100-45), 2 μg ml−1 doxycycline, 0.01 µg ml−1 IGF1 (PeproTech, 100-11), 0.05 μg ml−1 Laminin and 200 µM ascorbic acid (Thomas Scientific, C988E92). This maturation medium is replaced every day for two consecutive days, and then half-media volume is replaced with fresh media every 2–3 days for 1 month.
Human MN–myotube co-culture in MFCsDay 2 differentiated myotubes were cultured in the distal part of MFCs coated with ECM (1:100) and maintained with induction medium as mentioned above.
For MN cultures, iPSC clones were cultured in six-well plates coated with Matrigel (Corning, 356234), grown in mTesr1 medium (STEMCELL Technologies, 85850) and passaged with mTesr1 medium containing 10 μM RI (Sigma-Aldrich, Y0503) for 1 day after passaging. Culture media were refreshed daily until colonies reached 80% confluence. Doxycyline-induced differentiation into lower MNs was performed as previously described with minor modifications. In total, 300,000 iPSCs were plated in a 35-mm dish in mTesr1-RI medium. On the following day, media were replaced with induction medium supplement containing DMEM/F12 (Gibco, 31330038), 1% N-2 Supplement (Gibco, 17502048), 1% NEAA (Biological Industries, 01-340-1B) and 1% GlutaMAX (Gibco, 35050038) with 10 μM RI, 2 μg ml−1 doxycycline (Sigma-Aldrich, D9891) and 0.2 μM Compound E (Merck, 565790).
After day 9, day 2 differentiated MN cells (48 hours after doxycycline induction) were resuspended with Accutase (Sigma-Aldrich, SCR005) and re-plated in the proximal compartment of MFCs at a concentration of 50,000 MNs per MFC. Prior to plating, MFCs were coated overnight with 0.1 mg ml−1 PDL (Sigma-Aldrich, P6407) in PBS and 15 μg ml−1 Laminin (Sigma-Aldrich, L2020) for 4 hours on the following day. To prevent outgrowth of mitotically active cells, 40 μM BrdU (Sigma-Aldrich, B9285) was added to the medium during the first 24 hours after plating. At the fourth day, cells were treated with MM medium containing the following: Neurobasal medium (Gibco, 21103049), 1% B27 (Gibco, 17504044), 1% N-2, 1% NEAA, 1% Optimal-Culture-One supplement (Gibco, A3320201), 1 µg ml−1 Laminin, 20 ng ml−1 BDNF, 20 ng ml−1 GDNF and 10 ng ml−1 NT3 (Alomone Labs, N-260). Medium was refreshed every 3 days.
MN crossing into the myotube compartment was observed usually within 3–4 days after culturing. Throughout the co-culture condition, separate media components for MNs and myotubes were followed as aforementioned. For imaging, chambers were fixed using 4% PFA for 15 minutes and then processed for immunofluorescence protocol.
Automated image analyses using CellProfilerAlong with ImageJ, CellProfiler 4 was used to build an unbiased semi-automated image analysis to perform quantitation of percentage of degenerated neurons (neurofilament heavy chain (NFH) staining) and amount of axonal pTDP-43 in MN–myotube co-culture of isogenic and ALS lines. Deconvolution of z-stacked images is performed using ImageJ where each slice from each fluorescent channel is exported as a TIFF file. To begin quantification, the images to be quantified must first be loaded into the input panel. For both NFH degeneration and axonal pTDP-43 analysis, maximum intensity projections of NFH and pTDP-43 channel images were loaded into the pipeline module. Once an image has been loaded into the pipeline, the image will be converted into separate grayscale images representing each of the color channels using the ‘ColorToGray’ module. To enhance neurite particles within images, we use the ‘EnhanceOrSuppressFeatures’ module to apply filtering and opt to enhance the identification of neurites. To identify ‘intact NFH’, we use the ‘IdentifyPrimaryObjects’ module using the grayscale maximum intensity of NFH channel image as the input image. Within the ‘IdentifyPrimaryObjects’ module, we selected ‘Global Otsu Thresholding’ using two-class thresholding, set typical diameter of objects between 10 and 120 pixel units and used both ‘intensity’ and ‘shape’ features to differentiate clumped objects. To identify ‘degenerated NFH’ objects from the same NFH channel, we used the same method of ‘IdentifyPrimaryObjects’ module of same thresholding and set the typical diameter of objects between 3 and 8 pixel units. For axonal pTDP-43 quantification, a separate image analysis pipeline was generated with additional modules. In addition to the initial steps of ‘ColorToGray’ module and ‘IdentifyPrimaryObjects’ module to identify NFH and pTDP-43 from their image channels, additional modules such as ‘RelateObjects’ and ‘MaskObjects’ were used to identify axonal pTDP-43 co-localization. To identify the individual pTDP-43 particles within each NFH image, we used ‘NFH’ objects as the parent objects and ‘pTDP-43’ objects as the child objects within the module. Ultimately, this allows for the counting of ‘pTDP43 objects’ per ‘NFH objects’. Lastly, all the data collected were exported into separate spreadsheets using the ‘ExportToSpreadsheet’ module that is provided with counts of particles per image in .csv format. From this, the ratio of particle count of degenerated NFH to intact NFH is used to calculate the percent of degenerated axons in each co-culture, and the ratio of number of pTDP43 objects present in each NFH object is used to calculate the amount of axonal pTDP-43.
Immunofluorescent staining of cryosectionsSciatic nerve and spinal cord sections were prepared from fixating respective tissues in 4% PFA for 16 hours at 4 °C and then incubated with 20% sucrose for 16 hours at 4 °C and cryo-embedding in Tissue-Tek OCT compound (SciGen). Tissues were then cryosectioned to 10-µm-thick slices and washed with PBS, followed by permeabilization and blocking in solution containing 10% goat serum, 1 mg ml−1 BSA and 0.1% Triton in PBS for 1 hour. Later, the sections were incubated overnight at 4 °C with primary antibodies: rabbit anti-TDP-43 (1:2,000; Proteintech, 10782-2-AP), rabbit anti-pTDP-43 (1:2,000, Proteintech, 22309-1-AP), chicken anti-NFH (1:500; Abcam, ab72996) and mouse anti-S100 B (1:300; Sigma-Aldrich, S2532). This was followed by 2-hour incubation at room temperature with secondary antibodies: goat anti-chicken 405 (1:500; Abcam, ab175675), goat anti-chicken 594 (1:500; Thermo Fisher Scientific, A-11042), goat anti-mouse 488 (1:500; Abcam, ab150113) and goat anti-rabbit (1:500; Abcam, ab150083). Samples were washed with PBS and mounted with ProLong Gold (Life Technologies).
Immunofluorescent labeling of cell culturesCell cultures (either mass cultures or in MFCs) were performed as previously described. In brief, cultures were fixed for 15 minutes with 4% PFA at room temperature, followed by consecutive PBS washes. For co-cultures, labeling of postsynaptic AChRs was performed prior to permeabilization by incubation with either BTX-ATTO-633 or TMR-BTX (0.5–1 mg ml−1 in PBS) for 15 minutes at room temperature. After PBS wash, cultures were permeabilized for 30 minutes at room temperature with 0.1% Triton X-100 in PBS or with permeabilization/blocking solution (0.1% Triton X-100, 5% goat serum and 1 mg ml−1 BSA in PBS) for 30 minutes. After PBS rinse, cultures were incubated for 1 hour with blocking solution (5% goat serum and 1 mg ml−1 BSA in PBS), following overnight incubation at 4 °C with primary antibodies in blocking solution. After PBS rinse, cultures were incubated for 2 hours at room temperature with secondary antibodies in blocking solution, followed by several PBS washes.
Samples were mounted with ProLong Gold antifade reagent (±DAPI; Molecular Probes). In some experiments, samples were labeled with Phalloidin-FITC (1:250) after permeabilization (and other relevant labeling procedures) to mark axons instead of immunofluorescent labeling. Primary antibodies used included: rabbit anti-TDP-43 (1:2,000; Proteintech, 10782-2-AP), rabbit anti-pTDP-43 (1:2,000; Proteintech, 22309-1-AP), chicken anti-NFH (1:500; Abcam, ab72996), rabbit anti-NFH (1:1,000; Sigma-Aldrich, N4142), rabbit anti-Rab27a (1:200; Proteintech, 17817-1-AP), chicken anti-GFP (1:500; Abcam, ab13970), rabbit anti-Ago2 (1:500; Abcam, ab32381) and mouse anti-Titin (1:500; DSHB, 9D10). Secondary antibodies used included: goat anti-chicken 405 (1:1,000; Abcam, ab175675), goat anti-rabbit 405 (1:500; Abcam, ab175654), goat anti-chicken 488 (1:1,000; Abcam, ab150173), goat anti-rabbit 594 (1:1,000; The Jackson Laboratory, 111-585-144), goat anti-rabbit 488 (1:1,000; Invitrogen, A11034), goat anti-mouse 488 (1:500; Abcam, ab150113), goat anti-mouse 594 (1:1,000; Invitrogen, A11032) and goat anti-rabbit 647 (1:500; Abcam, ab150083).
Whole-mount NMJ stainingGastrocnemius, tibialis anterior, EDL and soleus muscles were dissected from adult mice, cleared from connective tissue and kept in 4% PFA until use. Muscles were washed in PBS, dissected into small muscle bundles (approximately 200 fibers per bundle) and stained for postsynaptic AChR with αBTX-ATTO-633 (Alomone Labs) or αBTX-TMR-594 (Sigma-Aldrich) at 1 µg ml−1 for 20 minutes at room temperature while rocking. Next, muscles were permeabilized with ice-cold MeOH at −20 °C for 5 minutes, blocked and further permeabilized with 20 mg ml−1 BSA and 0.4% Triton for 1 hour. Muscle preparations were agitated overnight at room temperature with appropriate antibodies: rabbit anti-TDP-43 (1:2,000; Proteintech, 10782-2-AP), rabbit anti-pTDP-43 (1:2,000; Proteintech, 22309-1-AP), chicken anti-NFH (1:500; Abcam, ab72996), rabbit anti-Rab27a (1:200; Proteintech, 17817-1-AP), mouse anti-Synapsin I (1:500; Millipore, AB1543), guinea pig anti-Synaptophysin (1:300; SYSY, 101004), CD63 (1:200; Santa Cruz Biotechnology, H-193, sc-15363), rabbit anti-CD9 (1:100; Abcam, ab92726), mouse anti-CD81 (1:100; Abcam, ab79559), rabbit anti-CHMP2a (1:500; Proteintech, 10477-1-AP) and chicken anti-GFP (1:500; Abcam, ab13970). This was followed by 4-hour incubation at room temperature with secondary antibodies: goat anti-chicken 405 (1:500; Abcam, ab175675), goat anti-chicken 488 (1:500; Abcam, ab150173), goat anti-mouse 488 (1:500; Abcam, ab150113), goat anti-rabbit 647 (1:500; Abcam, ab150083) and goat anti-guinea pig-488 (1:500; Abcam, ab150185). In case required, DAPI was diluted within PBS used for the secondary washes, incubated with the samples for 5 minutes and then washed. Finally, muscle bundles were cut and spread into thinner layers and mounted with VECTASHIELD (Vector Laboratories). Cover slides were sealed with nail polish until use.
Dissociation of muscle fibers prior to their labeling was done by incubating freshly isolated gastrocnemius muscle in DMEM (4.5 g l−1 glucose) containing 2.5% penicillin–streptomycin–nystatin (PSN, v/v; Biological Industries) and Collagenase I (2 mg ml−1; Sigma-Aldrich, C-0130) for 3 hours at 37 °C and 5% CO2. Afterwards, muscles were transferred into a BSA-coated (5% w/v in ultrapure water) 100-mm dish with 15 ml of DMEM (4.5 g l−1 glucose) containing 2.5% PSN and left in the CO2 incubator for 30 minutes. Lastly, muscles were dissociated into single fibers by gradual trituration using a BSA-coated 1-ml tip (pre-cut to have a wider opening). Fibers were collected into a 1.7-ml Eppendorf tube using a fire-polished BSA-coated Pasteur pipette and briefly spun down, and medium was replaced with fresh 4% PFA. Dissociated muscle fibers were labeled similarly. To replace the various reagents during the staining procedure, fibers were gently spun down in each step for discarding maximal volume of the used solution.
miR-126-expressing viral particlesGenes of interest were cloned into a third-generation lentiviral pLL3.7 backbone with either cytomegalovirus (CMV) or hSynapsin promoters. HEK293T cells were transfected by employing the calcium phosphate method and a mixture consisting of the vector of interest and vesicular stomatitis virus glycoprotein, and group antigens–polymerase (reverse transcriptase) was used. The medium was replaced after 5–8 hours, and the supernatant was collected 48 hours later. Next, 50 mM HEPES was added before freezing to maintain a neutral pH for long-term storage at −80 °C. If necessary, the lentiviruses were concentrated using a PEG Virus Precipitation Kit (Abcam). Lastly, the lentiviruses were stored in Neurobasal medium (Gibco) at −80 °C. Lentiviruses were used to overexpress miR126-5p in muscle and MN primary cultures. Two hours after plating, the cells were infected with pLL-miR126-5p–GFP or with pLL–GFP (106 titer units). Custom-made single-stranded AAV vectors, serotype 9 variant PHP.eB expressing miR-126 or GFP, were designed and manufactured by the Viral Vector Facility of ETH Zurich. The titer of the viruses was verified by RT–qPCR in the facility and got virus concentration of approximately 3.2 × 1013 viral genomes per 1 ml.
AAV- PHP.eB miR-126 delivery to miceThe injection procedure was performed on presymptomatic approximately P60 mice. Mice were first anesthetized using a mixture of xylazin and ketamine. Then, double injection was performed: intramuscular injection to the right gastrocnemius and retro-orbital injection to the right eye. Each injection was of 25 µl of AAV.PHP.eB–GFP or AAV.PHP.eB miR126-5p–GFP (total of 50 µl per mouse, equal to 1.6 × 1012 viral genomes) using a 25-µl Hamilton syringe and a 30-gauge Hamilton needle. All animal experimentations were approved by the Tel Aviv University Animal Ethics Committee.
Gait analysis and phenotypic characterization of live animalsEvaluation of the motor abilities in SOD1G93A and healthy mice delivered with AAV.PHP.eB particles was performed using the CatWalk XT gait analysis platform (Noldus Information Technology). The test was done at the ages of P90 (early symptoms) and P130 (late symptoms), 30 days and 70 days after injections, respectively. Moreover, mice weight and survival were monitored throughout the entire experiment duration. Experiment endpoint was set at the point of which mice had reached endstage disease condition at neuronal score 5 or at P200, after which tissues were isolated for further analyses, such as immunofluorescence and PCR.
RNA extraction and cDNA synthesisMN axonal RNA was extracted from the outer compartment of radial MFCs at 14 days in vitro (DIV). Axonal RNA was extracted by removing the PBS (from prior wash) from the outer compartment and adding 100 µl of QIAzol lysis reagent (Qiagen). The inner well was filled with higher volume of PBS to disable the inward flow of lysis reagent toward the inner (soma) compartment and prevent soma contamination. Axons were washed off the plate by pipetting the QIAzol around the outer well for 30 seconds. RNA from somata in the inner compartment was extracted with 100 µl of QIAzol reagent, and lysate was collected in a similar manner. cDNA for axon and soma was prepared with High-Capacity Reverse Transcription Kit (Thermo Fisher Scientific, 4368814).
For sciatic nerve RNA extraction, sciatic axoplasm was obtained from two adult mice sciatic nerves in a tube containing 100 µl of RNase/DNase-free ultrapure PBS, cut into small pieces and gently squeezed on ice. The axoplasm was then centrifuged at 17,000g for 15 minutes at 4 °C. RNA was extracted using the RNAeasy Micro Kit (Qiagen) according to the manufacturer’s protocols, and RNA yield was quantified using a Qubit fluorometer (RNA Broad-Range Assay Kit; Thermo Fisher Scientific).
Exosomal RNA was extracted by dissolving the exosomal pellet in 700 µl of QIAzol.
RNA was extracted using the RNAeasy Micro Kit (Qiagen) according to the manufacturer’s protocols, and RNA yield was quantified using a Qubit fluorometer (RNA Broad-Range Assay Kit; Thermo Fisher Scientific). For standard whole-cell RNA preparations from MN/muscle cultures, cultures were lysed in 700 µl of TRI reagent, and RNA was isolated with ordinary chloroform, isopropanol precipitation protocol.
Small RNA next-generation sequencingFor small RNA next-generation sequencing, libraries were prepared from 10–15 ng of total RNA using the QIAseq miRNA Library Kit and the QIAseq miRNA NGS 48 Index IL (Qiagen), by an experimenter who was blinded to the identity of samples. Samples were randomly allocated to library preparation and sequencing in batches. Precise linear quantification of miRNA is achieved by using unique molecular identifiers (UMIs) of random 12 nucleotides after 3’ and 5’ adapter ligation, within the reverse transcription primers75. cDNA libraries were amplified by PCR for 19 cycles, with a 3’ primer that includes a six-nucleotide unique index, followed by on-bead size selection and cleaning. Library concentration was determined with a Qubit fluorometer (dsDNA High-Sensitivity Assay Kit; Thermo Fisher Scientific) and library size with TapeStation D1000 (Agilent). Libraries with different indices were multiplexed and sequenced on a NextSeq 500/550 version 2 flow cell or NovaSeq SP100 (Illumina), with 75-bp single read and 6-bp index read. FASTQ files were demultiplexed using the user-friendly transcriptome analysis pipeline (UTAP)76. Mouse miRNAs, as defined by miRBase77, were mapped using the Qiagen RNA-seq Analysis Portal 1.0 and 2.5.1, GeneGlobe pipeline (https://geneglobe.qiagen.com/us/analyze). We defined ‘true-positive’ miRNAs and reduced the likelihood of considering ‘false-positive’ miRNAs by including only miRNAs with an average UMI count >100 across all samples and with at least a single UMI across all samples, similar to our previous works78,79,80. Data were further normalized and average read counts compared between groups using the DESeq2 package81 under the assumption that miRNA counts followed negative binomial distribution. Significance was determined using the Wald test.
Axonal RNA-seqRNA was extracted from primary MN axons grown in radial MFCs and purified as described above13. RNA yields were quantified using a Qubit fluorometer (RNA Broad-Range Assay Kit; Thermo Fisher Scientific). RNA integrity was assessed by TapeStation. cDNA libraries were prepared from 10 ng of axonal RNA by the genomic unit in the Weizmann Institute of Science using the MARS-seq pipeline82. MARS-seq libraries with different UMIs were multiplexed and sequenced on a NovaSeq SP100 flow cell (Illumina), with 75-bp single read and 15-bp UMI read. After demultiplexing, FASTQ files were mapped to mouse genome using the GenCode annotations (vM25, 2020) in the UTAP piepeline76. We defined ‘true-positive’ genes and reduced the likelihood of considering ‘false-positive’ genes by including only genes with an average UMI count >15 across all samples. Data were further normalized and average read counts compared between groups using the DESeq2 package81 under the assumption that gene counts followed negative binomial distribution. Significance was determined using the Wald test.
RT–qPCR analysisSYBR Green-based assays (PerfeCTa SYBR Green FastMix; Quantabio) and StepOne Real-Time PCR system (Thermo Fisher Scientific) were used in RT–qPCR to quantify differential expression of mRNAs in MNs, MN axons and primary skeletal muscles. GAPDH mRNA was used as a reference RNA when calculating ∆CT. RT–qPCR primers and their sequences are listed in Supplementary Table 5.
Actb mRNA was used as a reference RNA when calculating ∆CT in Tardbp/Polb relative expression in axons and somata cDNA preparations.
PCR amplification and Southern blotReverse transcription was performed with the High-Capacity Reverse Transcription cDNA Kit using random primers (Thermo Fisher Scientific). Standard PCR was used to amplify sciatic axoplasm cDNA for detecting Tardbp, Polb and Actb mRNA using KAPA ReadyMix. Primer sequences are listed in Supplementary Table 5.
TaqMan miRNA RT–qPCRTaqMan RT–qPCR was done to evaluate the levels of mature miR-126-5p. U6 gene was used as a reference gene when calculating ∆CT, as we aim to quantify relative miR-126-5p levels in various tissues and cells.
For the TaqMan experiment, cDNA was extracted from the purified RNA using dedicated primers that will amplify miR126-5p signal using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368813). Later, we ran the samples in qPCR using the TaqMan method to quantify the alteration in miR-126-5p levels using miR126-5p and U6 advanced TaqMan primers (Thermo Fisher Scientific, 4427975). qPCR TaqMan reactions were performed with TaqMan Fast Advanced Master Mix for qPCR (Thermo Fisher Scientific) in a StepOne Real-Time PCR system (Thermo Fisher Scientific).
TDP-43 isoforms quantificationIllumina FASTQ files were uploaded for bioinformatics analysis to CLC Genomics Workbench version 24 (https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-clc-genomics-workbench/) and the ‘RNA-seq analysis’ tool with default options. In brief, reads were trimmed from 5’ adapter sequence of GTTCAGAGTTCTACAGTCCGACGATC. After this, trimmed reads were aligned to the GRCm39/mm39 mouse genome (downloaded from https://www.gencodegenes.org/mouse/) with the following parameters: mismatch cost of 2, insertion cost of 3, deletion cost of 3, length fraction of 0.8 and similarity fraction of 0.8. Aligned reads were annotated to mRNA tracks to define splicing isoforms per gene. Finally, isoforms were quantified by the CLC Genomics Workbench ‘expectation–maximization’ estimation algorithm.
Dual-luciferase reporter assayHEK293A cells were seeded a day before transfection in three 60-mm wells (2.5–3 × 105 per well; Thermo Fisher Scientific, 140675) per sample. Cells were co-transfected with mirVec plasmid expressing the pre-microRNA-126 and dual-luciferase (Firefly and Renilla Luciferase) plasmid (a kind gift from the Shomron laboratory, Tel Aviv University). Dual-luciferase plasmid was cloned downstream to Renilla Luciferase with 100-bp context sequence of the 3’ UTR of mmu-TDP-43 mRNA containing either the wild-type miR-126a-5p binding site or a mutant one where four nucleotides within this site have been mutated. Transfection was performed by Lipofectamine 2000 (Invitrogen, 11668-027) for 72 hours, after which cells were washed once with PBS (Sigma-Aldrich, D8537; 500 ml) and harvested according to the manufacturer’s protocol.
Serum EV purificationPatient blood serum samples were collected from Caucasian patients with ALS aged 45–88 years (68.37 ± 14.49) and healthy controls aged 23–66 years (43.87 ± 16.81) from both sexes (Supplementary Table 5), with collaboration of Amir Dori from Sheba Hospital. From each sample, 1 ml was used to purify EVs in order to evaluate the levels of mature miR-126-5p. Using the exoRNeasy kit (Qiagen, 76064), we isolated EVs from the serum samples and purified total RNA. The levels were normalized to the same volume and process for all the samples and conditions.
Protein extractionGastrocnemius muscles were extracted from adult mice and homogenized in ice-cold PBS lysis buffer containing 0.1% Triton X-100, protease and phosphatase inhibitors (Roche). Sciatic axoplasm was obtained from both sciatic nerves from every mouse. Sciatic nerves were sectioned, and axoplasm was extracted into 100 µl of PBS with protease and phosphate inhibitors by gentle pressing the sections. Proteins from primary MNs and primary muscle cultures were extracted using PBS lysis buffer containing 0.1% Triton X-100, protease and phosphatase inhibitors.
Tissue/culture lysates were centrifuged at 10,000g for 15 minutes at 4 °C. Protein concentration was determined using Bradford (Bio-Rad) or BCA (Cyanogen) colorimetric assays.
Western blottingProtein samples were mixed with SDS sample buffer and boiled at 95 °C for 10 minutes and then loaded to 10% acrylamide gels for SDS-PAGE. Proteins were transferred to nitrocellulose or PVDF membranes in buffer containing 20% MeOH. Membranes were blocked with 5% skim milk (BD Biosciences) or 5% BSA for 1 hour, followed by overnight incubation at 4 °C with primary antibodies: mouse anti-human-TDP-43 (1:2,000; Proteintech), rabbit anti-TDP-43 (1:2,000; Proteintech), rabbit anti-ERK1/2 (1:10,000; Sigma-Aldrich), mouse anti-α-tubulin (1:5,000; Abcam), rabbit anti-CD63 (1:100; Santa Cruz Biotechnology, H-193, sc-15363), rabbit anti-CD9 (1:100–1:1,000; Abcam, ab92726), mouse anti-CD81 (1:100–1:2,000; Abcam, ab79559), rabbit anti-Ago2 (1:500; Abcam, ab32381), rabbit anti-ALIX (1:250; Abcam, ab76608), mouse anti-SOD1 (1:500; Santa Cruz Biotechnology, FL-154), rabbit anti-Rab27a (1:200; Proteintech, 17817-1-AP) and GM130 (1:200; BD Biosciences, 610823), followed by species-specific HRP-conjugated secondary antibodies: rabbit 111-035-003 and mouse 715-035-151, both 1:10,000 (The Jackson Laboratory). Membranes were then washed with TBS with 0.05% Tween 20 (TBS-T) and incubated for 2 hours at room temperature with secondary HRP antibody (1:10,000; The Jackson Laboratoy), washed with TBS-T and visualized in an iBright 1500 ECL imager (Life Technologies) after 5-minute incubation with ECL reagents. Quantification was performed using FIJI ImageJ software version 2.3.0/1.53q.
MFC preparationMFCs were prepared as previously described13,30,83. In brief, PDMS (Dow Corning) was cast into custom-made epoxy replica molds, left to cure overnight at 60 °C, punched (6-mm/7-mm/9-mm punches according to the type of MFC), cleaned and positioned in 35-mm and 50-mm glass plates (WPI) or over 22 × 22-mm microscope cover glasses (Mariendfeld). Radial MFCs were positioned on pre-adhered round 13-mm cover glasses within 24-well plates.
MN culture and MN–myocyte co-cultureVentral spinal cords from embryonic day 12.5 (E12.5) embryos were dissected in HBSS prior to dissociation. For Rab27a shRNA experiments, co-cultures were performed with ventral spinal cord explants isolated as previously described29,30,83. In brief, ventral spinal cords were acquired transversely, sectioning spinal cords into small pieces after dissecting the dorsal horns and plating in MFC proximal compartment in Neurobasal (Gibco), 2% B27 (Thermo Fisher Scientific), 1% GlutaMAX (Gibco), 1% penicillin–streptomycin and 25 ng ml−1 BDNF (Alomone Labs).
Dissociated MN cultures were obtained by further trypsinization and trituration of explants. Supernatant was collected and centrifuged through BSA (Sigma-Aldrich) cushion. The pellet was then resuspended and centrifuged through an OptiPrep (Sigma-Aldrich) gradient (containing 10.4% OptiPrep, 10 mM tricine and 4% w/v glucose). MN-enriched fraction was collected from the interphase, resuspended and plated in the proximal MFC compartment at a concentration of 200,000 MNs per regular MFC and 250,000 per radial MFC. MNs were maintained in complete neurobasal medium containing Neurobasal, 4% B27, 2% horse serum (Biological Industries), 1% GlutaMAX, 1% penicillin–streptomycin, 25 µM β-mercaptoethanol, 25 ng ml−1 BDNF, 1 ng ml−1 GDNF (Alomone Labs) and 0.5 ng ml−1 CNTF (Alomone Labs). Glial cell proliferation was restricted by the addition of 1 µM cytosine arabinoside (Sigma-Aldrich) to culture medium in 1–3 DIV. At 3 DIV, BDNF concentration in proximal compartment was reduced (1 ng ml−1), whereas medium in distal compartment was enriched with GDNF and BDNF (25 ng ml−1) to direct axonal growth.
Myocyte culture was performed as previously described29. In brief, gastrocnemius muscles from a P60 adult C57BL/6J mouse were extracted into DMEM with 2.5% PSN (Biological Industries) with 2 mg ml−1 Collagenase I (Sigma-Aldrich) for 3 hours and then dissociated and incubated with BIOAMF-2 (Biological Industries) in Matrigel (BD Corning)-coated plates for 3 days. Myoblasts were purified by performing pre-plating for three consecutive days and then plated at a density of 75,000 per MFC. Remaining myocytes were cryopreserved in 95% FBS, 5% DMSO and were used according to need by rapid thawing at 37 °C and resuspension in pre-warmed BIOAMF-2 medium (without antibiotics), followed by 300g, 4-minute-long centrifugation. Pellet was resuspended in BIOAMF-2 medium (without antibiotics), and muscles were plated in Matrigel-precoated 24-well plates for overnight recovery (150,000 myoblasts per well). In case the experiment performed required transfection of plasmids (that is, Rab27a shRNA, CD63–GFP, CD63-pHluorin, PLKO.1), myocytes were transfected 8 hours after their plating in the 24-well plates (further details in the transfection method section).
Prior to their co-culturing with primary MNs, primary myocytes in 24-well plates were washed once with pre-warmed PBS and then detached with 100 -µl-per-well Trypsin C solution (Biological Industries) for 15 minutes and then collected in fresh pre-warmed BIOAMF-2 medium and processed through 300g, 4-minute-long centrifugation. Myocyte pellets were resuspended in BIOAMF-2 medium and plated in the neuromuscular compartment of an MFC 1 hour after primary MNs were plated in the neuronal compartment. To aid NMJ formation, media in all compartments were then replaced to poor neurobasal (PNB) medium, which contained only 1% penicillin–streptomycin and 1% GlutaMAX.
Vectors and primary muscle transfectionAs detailed above, transfection of genetic vectors into muscles was performed at the myocyte stage of differentiation and prior to their culturing in MFCs. Myocytes were transfected using 1 µg of DNA and 3 µl of TurboFect transfection reagent (Thermo Fisher Scientific) in 300 µl of Opti-MEM medium (Gibco). Transfection mix was prepared according to the manufacturerʼs instructions. Transfection mix was discarded after 12 hours, and myocytes were washed once with pre-warmed optimum medium and then left to recover for an additional 4 hours with BIOAMF-2 medium (no antibiotics). Muscles were then re-plated in MFCs as described previously.
CD63-peGFP C2 (plasmid no. 62964), pCMV-lyso-pHluorin (CD63-pHluorin; plasmid no. 70113) and pLKO-Tet-On-shRab27a (plasmid no. 120930) were acquired from Addgene repositories.
pLL3.7-Ago2-eGFP was constructed in the Tel Aviv University lentiviral facility. pLL3.7-TDP-43-eGFP was constructed by cloning TDP-43-eGFP into PLL3.7 backbone (acquired from Addgene).
For shRab27a experiments, doxycycline was applied exclusively at the neuromuscular compartment at 5 days into co-culture, in a concentration of 1 µg ml−1.
Live imaging of CD63-pHluorin signals was performed using Tyrode’s solution (2 mM CaCl2, 2.5 mM KCl, 119 mM NaCl, 2 mM MgCl2, 20 mM glucose and 25 mM HEPES (pH 7.4)) in the presence or absence of 50 mM NH4Cl (instead of NaCl) to neutralize intracellular pH.
Tracking muscle-derived RNA in axonsPrimary skeletal muscles were cultured and differentiated as mentioned above. Upon differentiation, SYTO RNASelect Green (5 µM; Invitrogen, S32703) was added to muscle growth media for 1 hour. Cultures were washed three consecutive times with PBS, and fresh collection media were added to cultures. Muscle conditioned media were collected for 3 hours and then centrifuged at 4,000g for 5 minutes and passed through a 0.45-µm filter. Medium was applied to the axons within the distal compartment of MFCs. Prior to conditioned media addition, neuronal RNA was labeled using SYTO64 (5 µM; Invitrogen, S11346) for 10 minutes and then washed away by consecutive PBS and medium washes.
GW4869 exosome/EV inhibitorMuscle experiments were conducted using GW4869 inhibitor (Sigma-Aldrich, D1692) dissolved in DMSO creating a 5 mM stock solution. The inhibitor or the DMSO control was diluted to the culture media (BIOAMF-2) to a final concentration of 10 µM.
In vitro NMJ quantificationNMJ disruption was determined by high-magnification confocal imaging and quantification of the overlap of presynaptic and postsynaptic markers at the neuromuscular compartment in co-cultures. At the endpoint of the experiments (normally 10–14 days), co-cultures in MFCs were fixed with 4% PFA for 15 minutes at room temperature. αBTX-ATTO Fluor-633 (1:200; Alomone Labs, B-100-FR) or TMR-αBTX (Sigma-Aldrich) was used to label the extracellular domain of postsynaptic nAchR prior to membrane permeabilization. The percentage of healthy NMJs was determined by dividing the number of intact co-localization events, with intact non-degenerated axons, by the total number of co-localization events identified in every MFC.
EV purification processEVs were isolated from culture media using an ultracentrifuge protocol as previously described. In brief, the conditioned medium from differentiated primary muscle cultures (7 DIV) grown in 10-cm plates was collected in serum-free conditions for 16 hours. Conditioned medium was then centrifuged at 300g for 10 minutes at 4 °C, and supernatant was collected and transferred to a new tube centrifuged at 2,000g for 20 minutes at 4 °C. Supernatant was collected again and centrifuged at 10,000g for 30 minutes at 4 °C. Supernatant was then transferred into ultracentrifugation suitable tubes (Ultra-Clear centrifuge tubes; Beckman Coulter, 344059) and centrifuged at 100,000g for 70 minutes at 4 °C using the Optima-80 XPN ultracentrifuge (Beckman Coulter) with SW41 Ti rotor. Supernatant was discarded, and EV pellet was washed with ultrapure PBS 1× and centrifuged again at 100,000g for 60 minutes at 4 °C. The purified EVs were resuspended with 35 µl of 3.5× Laemmli sample buffer for western blot, 30 µl of PBS 1× for electron microscopy, 50 µl of PBS 1× for functional experiments or 700 µl of QIAzol for downstream RNA isolation.
In addition to the standard ultracentrifugation procedure, EVs for western blots and NTA in Supplementary Fig. 3a,b, Fig. 5d,e and Fig. 5m,n were isolated from primary muscle cultures grown in two 35-mm dishes per condition (total 4 ml) after 48 hours of medium collection in serum-free medium containing DMEM (4.5 g l−1 glucose), GlutaMAX (1%), B27 supplement (2%) and NEAA (1%) and in the presence or absence of the relevant treatment or control. EVs were purified using exoRNAeasy Midi exosome purification kit (Qiagen, 77144), following the manufacturerʼs protocol. For the NanoSight NTA, isolated EVs were further diluted 10× in PBS (0.1 µm filtered). Sample preparation for SDS-PAGE was done by lysing the isolated EVs in 10× RIPA buffer (Millipore) containing protease and phosphatase inhibitor mix. EV protein extracts were further concentrated using Amicon 0.5-ml centrifugal filters (10-kDa molecular weight cutoff). Whole-cell lysates were prepared by washing the culture once with 1× PBS and lysing the cultures with 1× RIPA buffer containing protease and phosphatase inhibitor mix. Protein concentrations were determined by BCA, and equal amounts of protein were loaded.
TEM imaging and negative stainingSamples were adsorbed on formvar/carbon-coated grids and stained with 2% aqueous uranyl acetate. Samples were examined using a JEM 1400Plus transmission electron microscope (Jeol). Images were captured using SIS Megaview III and the iTEM imaging platform (Olympus). Images were analyzed using FIJI software version 2.3.0/1.53q. Preparation and imaging of the samples in TEM were conducted by Vered Holdengreber at Tel Aviv University.
Fluorescence microscopy and image analysisConfocal images were captured using a Nikon Ti microscope equipped with a Yokogawa CSU X-1 spinning disk and an Andor iXon897 EMCCD camera controlled by Andor IQ3 software version 3.0; an Andor BC-43 benchtop spinning disk confocal system controlled by Andor Fusion software version2.3a; or a 3i Marianas confocal system with a Zeiss Axio Observer 7 microscope, equipped with a Yokogawa CSU-W1 spinning disk and a Prime 95B Back-Illuminated Scientific CMOS camera controlled by SlideBook software version 6.0. Phase-contrast movies of muscle contraction were acquired using the same microscope. High-content imaging and analysis of phase neurite morphology was performed using Sartorius Incucyte S3, 2019A, including Neurotrack module (cat. no. 9600-0010). All live imaging experiments were performed with 5% CO2 and 37 °C humidified using an in situ microscope setup.
smFISHTDP-43 mRNA single-molecule inexpensive FISH (smiFISH) was performed as previously described13,84. In brief, primary MNs were isolated and cultured for 7 days on cover glasses. Labeling procedures were performed in an RNase-free environment and using RNAse-free reagents. Cultures were fixed with 4% PFA and permeabilized overnight with 70% ethanol. Before hybridization, 40 pmol of 27 oligo-mix probes were pre-hybridized to 50 pmol of the FLAP probe in 10 μl of 100 mM NaCl, 50 mM Tris-HCl and 10 mM MgCl2 (pH 7.9). Pre-hybridization was performed on a thermocycler using the following program: 85 °C for 3 minutes, 65 °C for 3 minutes and 25 °C for 5 minutes. Samples were incubated with SSC (Sigma-Aldrich, S6639)-based 15% formamide (Thermo Fisher Scientific, 17899) buffer for 15 minutes. Samples were hybridized overnight at 37 °C with 27× FLAP-Y-Cy-3-tagged complementary oligonucleotide probes targeting regions in mouse Tardbp transcript variant 3 or with 23× FLAP-Y-Cy-3-tagged complementary probes targeting mouse Actb (Integrated DNA Technologies; Supplementary Table 5). The final hybridization mixture contained the probe duplexes (2 μl per 100 μl of final volume), 1× SSC, 0.34 mg ml−1 tRNA (Sigma-Aldrich, 1753), 15% formamide, 2 mM VRC (Sigma-Aldrich, R3380), 0.2 mg ml−1 RNAse-free BSA (Roche, 10711454001) and 10% dextran sulfate (Sigma-Aldrich, D8906). Samples were washed twice with warm 15% formamide and 1× SSC buffer (1 hour each), followed by 30 minutes with 1× SSC buffer and 30 minutes with 1× PBS supplemented with Phalloidin-FITC (1:250). Prior to mounting, samples were labeled with DAPI (0.2 µg ml−1). Samples were mounted with VECTASHIELD antifade reagent.
Labeling of TDP-43 mRNA in NMJs in vivo was performed in a similar way with the following modifications. Freshly isolated EDL muscles were dissected into smaller fiber bundles and fixed in 4% PFA for 1 hour while rocking. After PFA washout, tissues were permeabilized for 1 hour with 1% Triton X-100 in diethylpyrocarbonate (DEPC) water while rocking. Samples were washed with DEPC-PBS and then pre-hybridized as mentioned above. smiFISH hybridization steps were performed similarly to cultured cell yet with continuous agitation. After post-hybridization washes as detailed above, tissues were fixed again with 4% PFA for 10 minutes, after which we performed standard whole-mount immunolabeled with chicken anti-NFH (1:500) overnight at room temperature while rocking. After primary antibody washout, samples were incubated with secondary Alexa Fluor-405 anti-chicken antibodies at room temperature for 4 hours while rocking. After secondary antibody washout, samples were mounted with VECTASHIELD antifade reagent.
TDP-43 puro-PLACombined TDP-43 and puro-PLA was performed as previously described28 in order to evaluate the extent of TDP-43 protein synthesis in MN axons. In brief, primary MNs were grown in MFCs in the presence or absence of primary skeletal muscles in the neuromuscular compartment. Puromycin labeling was performed exclusively to the distal (axonal or neuromuscular compartment) for 30 minutes at a concentration of 10 µM, followed by two rapid cold PBS 1× washes and fixation in 4% PFA for 15 minutes. To validate the specificity of puro-PLA labeling, protein synthesis was exclusively inhibited in the distal/neuromuscular compartment by the addition of anisomycin (40 µM) for 30 minutes prior to and along with puromycin labeling. To reduce the background labeling in neuromuscular co-cultures and to expose the presynaptic PLA signals, primary muscles were transfected with empty backbone vector (PLKO.1) expressing the puromycin resistance gene puromycin acetyltransferase (PAC) prior to their co-culturing
Comments (0)