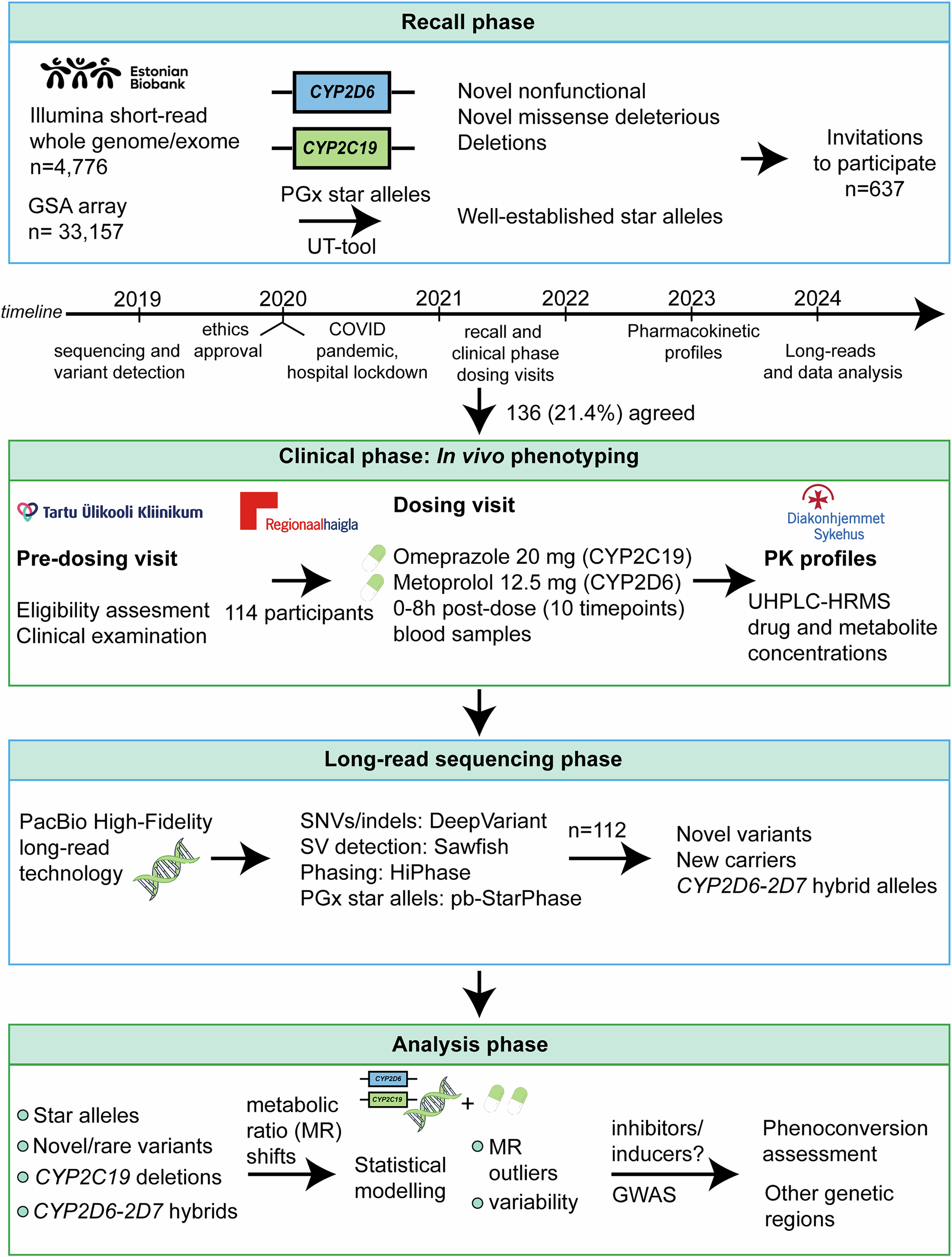
Variant detection and participant selection
Whole-genome and exome sequencing data obtained with Illumina short-read technology (n = 4776) were analysed for the participants of EstBB. The method for sequencing, variant calling, and filtering has been detailed in previous publications48,49,50,51. In this study, we identified all potential nonfunctional and missense variants in the CYP2C19 and CYP2D6 genes in the sequencing data, with a specific focus on novel variants. Variants that did not have a dbSNP identifier (rsID) at the time of recall were considered putatively novel.
Variant annotation and functionality predictions were performed using the Ensembl Variant Effect Predictor (version 87) and the LOFTEE plugin for identifying high-confidence nonfunctional variants. For missense variants, functional predictions were further refined using a prediction framework optimised for pharmacogenetic assessments52.
We used a previously developed star allele translation tool50—referred to in this study as the “UT-tool”—to assign pharmacogenetic star allele diplotypes for EstBB participants based on Illumina Global Screening array genotype data (n = 33,157) and Illumina short-read sequencing data of 4,776 individuals. Deletions were called using the PennCNV software53. Only samples with a standard deviation of log R ratio <0.3, absolute waviness factor <0.05, and number of copy number variant calls per sample <100 were retained for further analyses.
For recall, participants were invited based on the following selection criteria:
Having novel nonfunctional variants in CYP2C19 or CYP2D6;
Having novel missense variants predicted as deleterious;
Having the CYP2C19 deletions (CYP2C19*37, CYP2C19*42), including homozygous individuals and compound heterozygotes with different CYP2C19 star allele diplotypes;
Having CYP2D6 gene deletions or duplications;
Individuals with well-established reference allele, increased or no function alleles of CYP2C19 (*1, *2, *17) or CYP2D6 (*1, *4).
Eligible participants were adults (age ≥ 18 years, no upper age limit) who signed the informed consent form. Invitations were sent via email by EstBB personnel between 20/10/2021-23/08/2022. The study complied with all relevant ethical regulations including the Declaration of Helsinki.
Study visits and eligibility assessment
Responding participants (n = 174) underwent a pre-screening phone interview to confirm eligibility. Exclusion criteria included: Clinically relevant hepatic or renal dysfunction; anaemia; pregnancy or lactation; recent history (past year) of alcohol or drug abuse (self-report during clinical interview); use of medications known to interfere with probe drug metabolism; allergy or contraindication to omeprazole or metoprolol; body mass index (BMI) < 18 or >45.
Eligible subjects (n = 136) were invited for an in-clinic pre-dosing visit at Tartu University Hospital or North Estonia Medical Centre, where inclusion criteria were verified, informed consent obtained, and relevant clinical and laboratory assessments performed. Concomitant medications were reviewed to identify potential drug–drug interactions.
Probe drug administration and pharmacokinetic (PK) profile detection
During the probe drug dosing and PK sampling visit, eligibility was re-checked according to the study’s inclusion and exclusion criteria. A clinical examination was performed, and for fertile female participants, the absence of pregnancy was confirmed. Participants received a single oral dose of omeprazole 20 mg (commercially available capsule), metoprolol 12.5 mg (extemporaneously prepared capsule by Tartu University Hospital Pharmacy, as no commercial product was available for this dose). Participants were required to fast prior to probe drug administration to standardise PK conditions.
Blood samples (5 mL each) were collected at 10 time points: pre-dose (fasting) and at 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, and 8 hours post-dose. Samples were drawn from an antecubital vein via an indwelling cannula into EDTA tubes, centrifuged at 1500 g for 10 minutes at 4 °C, and plasma stored at −80 °C until analysis. The sampling schedule was based on prior cocktail phenotyping studies that showed 8-hour sampling adequately captures PK profiles in both normal and poor metabolisers38. The study subjects were recommended to have breakfast after the drawing of the 1 h blood sample. The composition of this meal was not standardised. Vital signs (blood pressure, heart rate) were monitored throughout the 8-hour visit. A follow-up phone call was conducted after one week to record any adverse drug reactions.
Plasma concentrations of probe drugs and their primary metabolites—omeprazole, 5-hydroxyomeprazole, 5-O-desmethylomeprazole; metoprolol, alpha-hydroxymetoprolol, O-demethylmetoprolol—were quantified using ultra high-performance liquid chromatography (UHPLC) and high-resolution mass spectrometry (HRMS). Samples were analysed at Center for Psychopharmacology, Diakonhjemmet Hospital, Oslo in Norway. Briefly, the plasma samples were prepared by protein precipitation in a semiautomated sample preparation procedure using a Microlab Star pipetting robot (Hamilton, Reno, NV, USA) for liquid handling. The LC system was a Vanquish-UHPLC (Thermo Fisher Scientific, Waltham, MA, USA), and chromatographic separation was performed by an XBridge BEH C18-column (2.6 μm, 2.1 ×75 mm; Waters, Milford, MA, USA) using gradient elution at 35°C with a mix of ammonium bicarbonate buffer (pH = 8.1) and methanol (43–60%). The injection volume was 4 µL and total run time was 2.6 min.
Detection used a QExactive Orbitrap mass spectrometer (Thermo Fisher Scientific) operated in positive ionization mode, acquiring full-scan data at a resolution of 70,000 within the 100- to 1500-Da scan range. The compounds were quantified in full-scan acquisition mode, whereas accurate data-dependent MS2 analysis was simultaneously triggered to permit confirmation of their identification. Within-run and between run CVs and accuracies were <7% and 93–106% for all compounds at the low-quality control level (50 nM for metoprolol and metabolites, and 100 nM for omeprazole and metabolites).
Measures of enzyme activity
Drug and metabolite concentration measurements were imputed as zero if the analyte was not detected and as LOD/2 if the concentration was below the limit of detection (LOD = 0.4 nM for omeprazole and its metabolites; LOD = 0.2 nM for metoprolol and its metabolites). PK analysis was performed using non-compartmental analysis. The area under the curve (AUC) from time zero until the last concentration measurement was determined for each analyte using the log-linear trapezoidal rule (linear-up log-down method) with the R package pkr. The metabolic ratios were computed as the AUC of the parent drug divided by the AUC of its metabolite to reflect enzyme activity.
The two metabolic ratios calculated for reflecting the CYP2C19 enzyme activity—AUC of omeprazole-to-5-hydroxyomeprazole and -5-O-desmethylomeprazole—were highly correlated (Pearson correlation 0.95). Therefore, we are reporting the results using the AUC of omeprazole-to-5-hydroxyomeprazole only. The metabolic ratio using the AUC of metoprolol-to-alpha-hydroxymetoprolol was used for reflecting the CYP2D6 enzyme activity, because the metabolic ratio using O-demethylmetoprolol was less effective in separating the different groups of CYP2D6 metabolisers. The metabolic ratios were log-transformed in all statistical analyses.
CYP2C19 deletion frequency in the Estonian Biobank
To estimate the frequency of the CYP2C19 partial gene deletion (CYP2C19*37 or CYP2C19*42) in EstBB, individuals with deletion were identified using PennCNV53 applied to genotyping data from the Illumina Global Screening Array in 17 batches. Duplicates and samples with call-rate <0.95 were excluded. We only considered deletion calls that (i) fell into the boundaries of an established 61.8k deletion overlapping CYP2C19 exons 1 to 5 (gnomAD structural variants v4.1.0 variant ID: DEL_CHR10_28B50744)31, and (ii) were at least 5k base pairs long. All other CYP2C19-overlapping deletions were flagged as ambiguous.
Post-recruitment star allele re-assignment with new tools
Since pharmacogenetic star allele calling tools keep evolving, we performed additional star allele assignments for a subset of participants (n = 43) who had short-read whole-genome sequencing data available. We determined star allele diplotypes for CYP2D6 using two specialised freely available computational tools: Cyrius v1.1.154 and Aldy v4.555, using default parameters and the GRCh38 reference genome in the calling process.
The diplotype results from these tools were compared against two benchmarks: the UT-tool50, which was used for star allele calling during the recruitment phase, and pb-StarPhase46, which we consider the analytical gold standard in this study due to its integration of long-read sequencing, phasing, and CYP2D6-D7 specific reference sequences for accurate alignment and star allele calling46. The UT-tool reports diplotypes based on the set of defined CYP2C19 and CYP2D6 star alleles in the tool database and does not detect or assign novel SNPs or previously uncharacterized haplotypes. For CYP2C19, we expanded the allele calling to include all 114 participants by applying the PharmCAT algorithm56 on phased genotype data derived from both microarrays and sequencing. The obtained CYP2C19 star allele assignments were then systematically compared with the calls produced by the UT-tool and pb-StarPhase to assess general concordance. All concordance analyses and comparative evaluations were performed using R (version 4.4.3)57, with custom scripts developed to calculate match rates.
Post-recruitment long-read sequencing of participants
A comprehensive long-read sequencing approach enables high-confidence characterisation of both SNVs and structural variants in CYP2C19 and CYP2D6, particularly for complex or rare haplotypes that are not reliably resolved by short-read sequencing. To resolve hybrid gene variants (e.g., CYP2D6-CYP2D7 hybrids), confirm suspected structural rearrangements, and refine star allele configurations, we performed long-read sequencing on 112 study participants. Sequencing was not conducted for two individuals (star allele diplotypes are *1/*1 for both genes based on the UT-tool, one individual has a CYP2C19 deletion predicted from genotyping data using PennCNV).
Genomic DNA extracted from peripheral blood samples was sequenced using PacBio Revio sequencing technology to generate highly accurate circular consensus HiFi (High-Fidelity) reads. Library preparation and sequencing were performed according to the manufacturer’s standard protocols. All samples (n = 112) were sequenced at an aimed coverage of 20X (mean = 23.7, median = 21.7). For each sample, we required a minimum of 57.5 Gbs of raw unmapped sequence to be processed further. HiFi reads were aligned to the human reference genome (GRCh38/hg38) using pbmm2 (v1.17.0). SNVs and small insertions/deletions (indels) were called using DeepVariant (v1.6.1) with the PacBio HiFi-specific model. Haplotype phasing was carried out using HiPhase58 (v1.4.5). SNVs and indels were functionally annotated using the Ensembl Variant Effect Predictor (VEP, v112), with plugins including dbSNP and gnomAD for functional classification and population frequency assessment. For structural variant detection, we employed sawfish (v0.12.10)59, a tool tailored for sensitive detection of deletions, duplications, and complex rearrangements from long-read data.
In this study, we focused on the interpretation of genetic variants specifically within the CYP2C19 region (chr10:94,762,681–94,855,547; GRCh38), which encompasses the full coding sequence. The well-known upstream regulatory variant CYP2C19*17 (c.-806C>T, rs12248560; chr10:94,761,900) was added to the analysis. For CYP2D6, we only analysed the coding region chr22:42,126,499–42,130,865. All variants with PASS filter and QC > 20 were further studied. Variants that did not have a dbSNP identifier (rsID) were considered putatively novel and were further evaluated for their functional significance. This evaluation included predicted consequences such as no function or missense variants. In addition, we examined which of the detected variants are currently listed in PharmVar30 (CYP2C19 and CYP2D6 haplotype GRCh38 files, accessed 21/03/2025). We assessed the impact of variants not covered by PharmVar, specifically those predicted to be nonfunctional or missense, on metabolic ratios.
Long-read-derived star allele calls
We assigned CYP2C19 and CYP2D6 star allele diplotypes using the pb-StarPhase tool (version v1.3.2)46, applying a minimum read support threshold of 2. The long-read-derived star allele calls were compared with previously inferred diplotypes from Illumina short-read sequencing and genotyping array data, generated during the initial recruitment phase. For cases with discordant calls or complex SVs, we inspected and validated haplotype structures manually using Integrative Genomics Viewer. Star allele diplotypes were translated into predicted metabolic phenotypes using gene-specific Clinical Pharmacogenetics Implementation Consortium/ClinPGx allele-phenotype translation tables (accessed 18/03/2025). All diplotype-phenotype visualisations and comparison with in vivo metabolic ratios were performed using the R software57 (version 4.4.3).
Drug-drug interaction
Although the use of established CYP2C19 and CYP2D6 inhibitors or inducers was among the exclusion criteria during the recruitment phase, the long intervals between the first pre-dosing and second PK sampling visits (partly due to the COVID-19 pandemic) may have resulted in changes in medication use during this period. The presence of inhibitors and inducers was assessed using electronic health records of prescription medications purchased within the six months preceding the second study visit. The data on prescription drug purchases were extracted from digital drug prescription records of the Estonian Health Insurance Fund. The selection of inhibitors and inducers was based on The Drug Interaction Flockhart Table60, including substrates with strong, moderate and weak evidence for interactions.
To verify the use of purchased medications and to monitor potential unregistered use of the most important CYP2C19 and CYP2D6 inhibitors or inducers, primarily form the groups of centrally acting drugs, i.e. antidepressants (inhibitors) and antiepileptics (inducers), a plasma sample from each participant was analysed. This was done using a validated, multianalyte UHPLC-HRMS method applied for therapeutic drug monitoring of psychoactive drugs at Diakonhjemmet Hospital. The samples were prepared and analysed with the same instrumentation as described for the in vivo phenotyping measurements. The method identifies potential co-medications, including the enzyme-inhibiting antidepressants bupropion, fluoxetine, fluvoxamine and paroxetine, and the enzyme-inducing antiepileptics carbamazepine and phenytoin.
Statistical modelling
The effects of genotypes on metabolic ratios were investigated using linear regression. One member per related pairs (identity by decent > 20%) was excluded, resulting in a sample size of 113 individuals. Additionally, we removed CYP2D6 diplotypes present in only one individual. The metabolic ratios were log-transformed, and models were adjusted for covariates with significant effects (P < 0.05) identified through forward stepwise regression. The tested covariates included age, sex, BMI, weight, clinical measurements and CYP2D6 or CYP2C19 inhibitors use during the six months before the study visit. For omeprazole, the final models considered BMI and the use of CYP2C19 inhibitors six months prior to the study visit. For metoprolol, adjustments were made for the use of CYP2D6 inhibitors six months prior. Contrasts were applied to test the differences between the effects of genotypes other than normal metaboliser, defined as individuals with two normal function alleles, which serves as the reference level in the linear regression. R software57 (4.4.3) was used to conduct the analysis.
Genome-wide analysis of metoprolol and omeprazole log-transformed metabolic ratios was performed using long-read data with REGENIE v3.261, adjusting for sex, birth year and the first two genotype principal components. Individuals with non-European ancestry were excluded (n = 1), leaving 111 individuals for analysis. Regional association plots for the significant loci were generated using LocusZoom43.
List of abbreviations
AUC: Area Under the Curve; BMI: Body Mass Index; CYP: Cytochrome P450; CYP2C19: Cytochrome P450 family 2 subfamily C member 19; CYP2D6: Cytochrome P450 family 2 subfamily D member 6; DDI: Drug–Drug Interaction; EstBB: Estonian Biobank; GWAS: Genome-Wide Association Study; NMa: Normal Metaboliser Allele; PM: Poor Metaboliser; UM: Ultrarapid Metaboliser; RM: Rapid Metaboliser; IM: Intermediate Metaboliser; NM: Normal Metaboliser; PK: Pharmacokinetics; SNV: Single Nucleotide Variant; SV: Structural Variant; UHPLC: Ultra High-Performance Liquid Chromatography; HRMS: High-Resolution Mass Spectrometry; PharmVar: Pharmacogene Variation Consortium.


Comments (0)