
Remember me
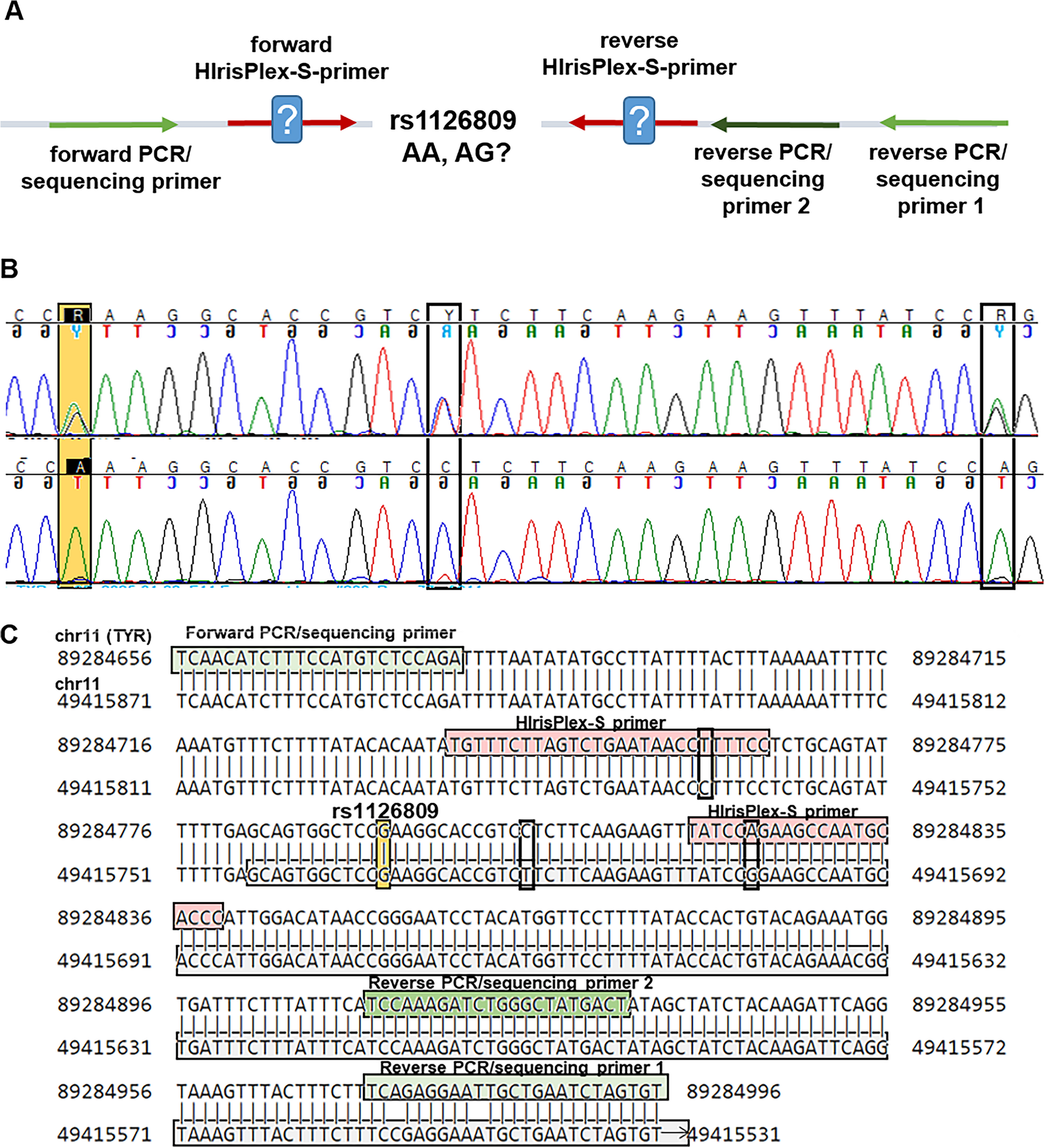


Foren-STR calculates both the allele and genotype frequencies for all loci from the input file and displays them in a table. These results can be viewed in the Description > Allelic Frequencies and Description > Genotypic Frequencies tabs. The allelic frequencies table is presented in a standard format and can be downloaded as a .TXT file or copy to clipboard. Similarly, the frequencies of all genotype combinations observed in the input file for all loci are displayed in a table and can also be downloaded same way.
To infer population substructure among different populations (Description > Heatmap tab), a heatmap based on the allelic frequencies of each locus in each subpopulation can be generated (see Fig. 1). This allows users to identify differences in allelic frequencies among subpopulations across all analyzed loci.
Fig. 1
Heatmap of allelic frequencies among three populations. A heatmap of the allelic frequencies of CSF1PO among three populations is plotted. The color depicts the allele frequency from 0 (blue) to 0.3 (red). In the y-axis is the name of the population and in the x-axis the alleles found in that marker. According to allele frequencies, population 1 and population 2 are more closely related than population 3
Forensic parametersThis software enables the calculation of common forensic parameters using an STR population database [4, 7]. The forensic parameters estimated by Foren-STR include Hardy-Weinberg equilibrium (HW), expected heterozygosity (He), and observed heterozygosity (Ho). Other calculations include the combined paternity index (PIC), typical paternity index (ITP), power of discrimination (PD), probability of exclusion (PE), and probability of match (PM). These calculations are performed per locus and combined, except for HW and PIC. The individual or combined results of these parameters can be presented by subpopulation or for the entire dataset in a table (Table statistics > Statistics tab), which can be downloaded as a TXT file or copy to clipboard.
Hardy Weinberg equilibriumThe Foren-STR software estimates whether the analyzed population is in Hardy-Weinberg (HW) equilibrium [6, 12]. This test is essential when calculating forensic parameters for an autosomal STR population database, as it identifies potential population stratification in the analyzed population. Foren-STR employs the same HW calculation method as the Arlequin software and in the pegas R package, using the following formula:
$$\:HW=\frac^_*!}^\prod\:_^_!}2H$$
Where n! is the factorial of the sample size, k is the number of different alleles in the analyzed locus, ni is the allele frequency of allele i, nij is the number of individuals with genotype i, j, 2n is the total number of alleles, and H is the frequency of heterozygotes for those alleles. This formula calculates the probabilities of all genotype combinations in the analyzed population. Using Markov-chain permutations and different contingency tables, the p-value of this calculation is estimated, allowing users to adjust for multiple comparisons, such as with Bonferroni’s correction.
Observed and expected heterozygosityThe observed heterozygosity (Ho) accounts for the proportion of heterozygous loci in individuals from a population. It is estimated by dividing the number of heterozygous individuals by the total number of individuals. On the other hand, the expected heterozygosity (He) is the probability that a locus is heterozygous and is calculated using the following formula:
$$\:He=\left[_^\left(1-_^_^\right)\right]/n$$
where n is the number of loci, m is the number of alleles found in locus j, and pij is the frequency of allele i in locus j.
Polymorphism information contentSimilar to heterozygosity, the polymorphism information content (PIC) measures the level of polymorphism present in a genetic marker. It is calculated using the following formula:
$$\:PIC=1-_^_^-\left(_^_^_^_^\right)$$
where n is the number of alleles, and pi and pj are the frequencies of alleles i and j. As the PIC value approaches 1, the genetic marker becomes more effective at discriminating between two unrelated individuals in a population.
Probability of matchThe probability of match (PM) is a common forensic parameter that estimates the probability of finding two identical genotypes in a population. It is calculated using the following formula:
Where pi is the frequency of genotype i at a specific locus in a population.
Power of discriminationThe power of discrimination (PD) is the probability that two unrelated individuals can be genetically distinguished by analyzing one or several genetic markers. It is calculated as follows:
Power of exclusionThe power of exclusion (PE) is the fraction of individuals who have a different DNA profile from a randomly selected individual in a paternity case. It is estimated using the following formula:
$$\:PE=^\left(1-2h^\right)$$
where h is the number of heterozygous individuals, and H is the proportion of homozygous individuals.
Typical paternity indexThe typical paternity index (ITP) estimates how many times more likely it is that the analyzed individual is the biological father compared to a random individual. This calculation is performed per locus using the following formula:
where H is the proportion of homozygous individuals.
Combined valuesTwo different approaches are used to estimate the combined values of forensic parameters. The combined values of Ho, He, PD, PM, and ITP are calculated by multiplying the individual values estimated for each marker. On the other hand, the combined values of PE and PD are calculated using the following formulas:
$$\:_=1-\left(1-_\right)\left(1-_\right)\left(1-_\right)\dots\:(1-_)$$
$$\:_=1-\left(1-_\right)\left(1-_\right)\left(1-_\right)\dots\:(1-_)$$
Genetic distancesWhen more than one population is analyzed, the Foren-STR software estimates the genetic distances between them using two approaches. The first is the standard Nei DA distance, which has been successfully used with microsatellite data [9]. The Nei DA distance is calculated as follows:
$$\:_=1-\frac_^_^_}\sqrt__}$$
where xij and yij are the frequencies of allele i at locus j in each population, mj is the number of alleles at locus j, and r is the number of analyzed loci.
The second method used to estimate genetic distances among populations using STRs is the Fst method proposed by [13], calculated as:
$$\:_=\frac_-_\right)/2-_}_}$$
Where J is the expected homozygosity for x and y populations.
Both values range from 0 to 1, with increasing values indicating greater genetic distance between populations. The results of these calculations are presented in an n×n matrix showing both DA and Fst values obtained from pairwise population comparisons (Table statistics > Distances tab). As with all estimations, these tables can be downloaded as .TXT files or copy to clipboard.
Comments (0)