Distinct Spatiotemporal Trajectories of Brain Volume in 3xTg-AD Mice: Rapid Entorhinal Atrophy and Early Hippocampal Growth Arrest
Identifying region-specific brain atrophy patterns is critical for early detection and intervention of AD. As an important region regulating learning and memory, MTL is known to be particularly vulnerable to early neurodegeneration (Chauveau et al. 2021). To systematically characterize these changes, we investigated global and region-specific brain volume changes in 3xTg-AD and WT mice across multiple ages.
We first quantified the whole-brain dimensions, including anteroposterior (AP) length, mediolateral (ML) width, and dorsoventral (DV) thickness, in WT and 3xTg-AD mice at 2, 5, 7, 12, and 18 months of age. Given the large workload of this comprehensive stereoscopic analysis across multiple ages, we initially characterized volume changes using male mice. In 3xTg-AD mice, all three dimensions followed a similar trajectory: a slight increase up to 7 months of age, reflecting normal postnatal brain development, followed by progressive shrinkage. By 18 months, all measures were significantly reduced in 3xTg-AD mice compared to WT controls (Fig. 1C, E, F), indicating a transition from early developmental growth to later neurodegeneration in overall brain size.
Next, we examined volume changes in the cerebral cortex and MTL subregions. Significant atrophy across the cerebral cortex emerged in 3xTg-AD mice at 12 and 18 months, indicating widespread degeneration in late AD stages (Fig. 1G and S1A-B). Notably, MTL subregions showed distinct temporal patterns of volume loss. In 3xTg-AD mice, the Ent showed the earliest and most severe atrophy, with significant reduction detectable by 7 months of age and progressive decline thereafter (Figs. 1H and 6.17 mm3 in 3xTg-AD vs. 7.25 mm3 in WT; 14.9% decrease, P = 0.0036). In contrast, the perirhinal cortex (PrC) and postrhinal cortex (POR) exhibited delayed atrophy, becoming significant only at 12 and 18 months (Fig. 1I, J). These data suggest that neurodegeneration spreads sequentially from the Ent to adjacent cortical areas as AD progresses.
Hippocampal volume also displayed distinct developmental trajectories between genotypes. In WT mice, hippocampal volume increased steadily during early postnatal stages and stabilized thereafter, indicating normal postnatal development (Hammelrath et al. 2016; Zhang et al. 2005; Calabrese et al. 2013). In 3xTg-AD mice, however, hippocampal growth was impaired early in development, leading to significantly smaller volume by 5 months of age relative to WT controls (Figs. 1K and 16.54 mm3 in 3xTg-AD vs. 18.99 mm3 in WT; 12.9% decrease, P = 0.0119). These findings imply that the smaller hippocampal volume in 3xTg-AD mice stems from an early neurodevelopmental growth arrest, rather than from the progressive degenerative atrophy seen in the Ent.
Beyond the MTL, significant volume loss was also observed in the PFC, a region important for cognitive regulation (Salat et al. 2001). However, different from the Ent, PFC atrophy in 3xTg-AD mice emerged later, with significant reductions detected only at 18 months of age (Fig. 1L and S1C).
To conclude, morphological analysis of 3xTg-AD brains revealed distinct region-specific volume changes during AD progression. The Ent showed rapid progressive atrophy, while the HPC exhibited early growth arrest (Fig. 1M). These findings suggest that the AD pathogenesis in this model may involve not only progressive neurodegeneration, but also early neurodevelopmental defects. Furthermore, the late-stage atrophy observed in multiple brain regions (PrC, POR, PFC) indicates progressive disease spread throughout the brain.
Early Excitatory Neuron Loss and Distinct Glial Alteration Underlie Entorhinal Vulnerability in 3xTg-AD Mice
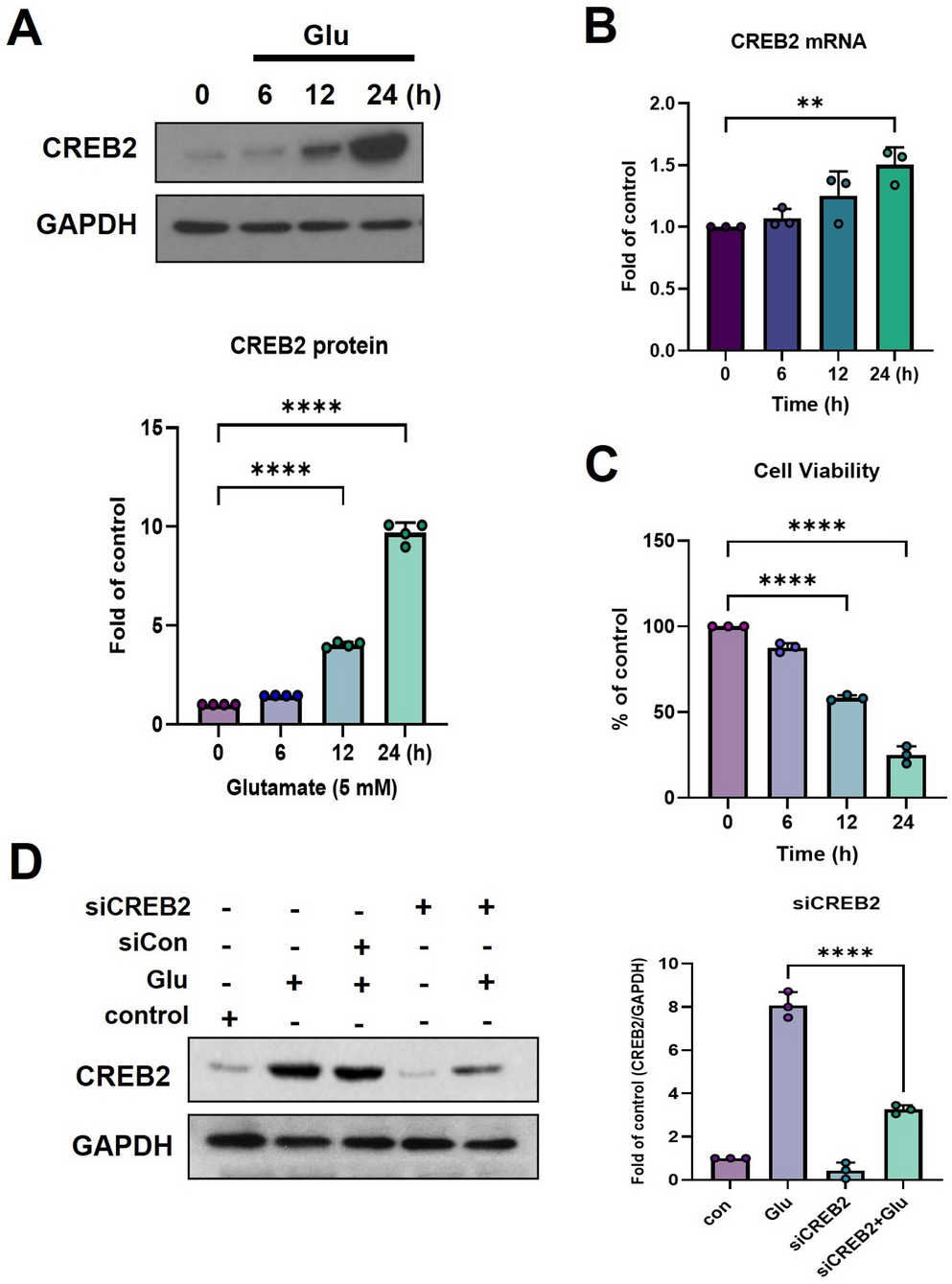
To further investigate the cellular basis of Ent atrophy, we analyzed neuronal density, cell-type-specific apoptosis, and glial activation within this region. Histological analysis revealed progressive neuronal degeneration in the Ent of 3xTg-AD mice. Overall neuronal density declined with age and was significantly lower in 3xTg-AD mice than in age-matched WT mice by 7 months of age (Fig. 2AA, 1.34 × 105 vs. 1.57 × 105 cells/mm3, P = 0.0278). Layer-specific analysis further demonstrated that this neuronal loss was most pronounced in layer II of the Ent, which exhibited a 17.4% reduction in density compared to WT controls (Figure S2 A-B, 2.06 × 105 vs. 2.49 × 105 cells/mm3, P = 0.0237). By later stages of AD, neuronal degeneration extended to all Ent layers. In parallel, the average neuronal soma size progressively decreased (Fig. 2B), and frequency distribution analysis revealed a shift toward smaller soma profiles (Figure S2C), suggesting continuous neuronal shrinkage. Cell-type-specific analysis via RNA in situ hybridization further revealed preferential reduction in excitatory neurons in the Ent of 3xTg-AD mice at 7 months, with a 17.2% decrease relative to WT controls (Figs. 2D, top, and 432.3 vs. 521.8 cells/mm2, P = 0.0472). Inhibitory neurons remained unchanged at this stage. By later time points, however, both excitatory and inhibitory neuronal populations were substantially diminished, reflecting widespread neuronal network disruption (Fig. 2D).
Glial cells in the Ent also exhibited distinct, age-dependent responses. Astrocytes displayed early and persistent atrophy, with significantly reduced somatic volume in 3xTg-AD as early as 2 months of age (Figs. 2F and 143.0 µm3 in 3xTg-AD vs. 230.6 µm3 in WT; 38.0% decrease, P < 0.001), a pattern consistent across all Ent layers (Figure S3B). Microglia, in contrast, underwent a gradual activation process (Fig. 2H-I). A growing proportion adopted a reactive phenotype by 5 months of age, characterized by enlarged somata and shortened, thickened processes (Figs. 2I and 14.25% in 3xTg-AD vs. 35.95% in WT; P = 0.017). By later stages (12 and 18 months of age), most microglia adopted a fully activated amoeboid morphology. Although astrocyte and microglial density in the Ent remained unchanged across ages (Fig. 2G and J), the absolute cell number likely decreased in proportion to overall tissue atrophy. In 3xTg-AD mice with established pathology, we also assessed glial associations with Aβ plaques and phosphorylated tau (pTau) tangles (Fig. 2K). Quantification of these interactions revealed a clear divergence in glial responses: as early as 7 months of age, microglia were found near the vast majority (86.7%) of aggregates, whereas astrocytes were associated with significantly fewer (44.0%) aggregates (P < 0.001; Fig. 2L). This profound disparity persisted at 12 and 18 months of age. Notably, entorhinal astrocytes remained distant from these aggregates even at advanced disease stages, indicating a limited reactive response, while microglia were frequently localized near Aβ and pTau deposits, consistent with a role in active immune surveillance.
Taken together, these results identify the Ent as an early and critically vulnerable site in the 3xTg-AD mouse model. In this model, the pathogenesis involves not only progressive neurodegeneration, as exemplified by the rapid atrophy of the Ent, but also early neurodevelopmental defects, as indicated by the hippocampal growth arrest. The Ent is characterized by early selective excitatory neuronal loss and a distinct glial response, wherein sustained astrocytic atrophy likely contributes to limited anti-inflammatory effects, thereby exacerbating local circuit dysfunction.
Early Neurogenesis Deficits and Distinct Glial Activation Characterize Hippocampal Pathology in 3xTg-AD Mice
The HPC, a key region for learning and memory, exhibits a different pathological trajectory from that of the Ent in 3xTg-AD mice (Fig. 1). We further investigated the specific vulnerability of the HPC in this AD model.
Neurogenesis in the dentate gyrus (DG) is essential for memory processes (Mu and Gage 2011; Hainmueller and Bartos 2020). Using doublecortin (DCX) as a marker of immature neurons in the DG, we found that neurogenesis was significantly impaired in 3xTg-AD mice as early as 2 months of age. DCX-positive cell density in the DG was 46.1% lower in 2-month-old 3xTg-AD mice than in WT controls (Figs. 3A-B and 223.0 vs. 120.3 cells/mm2, P < 0.001). By 7 months of age, DCX-positive cells were nearly absent in 3xTg-AD mice, indicating the cessation of neurogenesis in the DG.
Analysis of hippocampal subregions revealed an age-dependent loss of NeuN-positive neurons in the CA1 and CA3 of 3xTg-AD mice, which became statistically significant by 18 months of age (Figure S4 A-B, 1.85 × 105 vs. 1.55 × 105 cells/mm3, P = 0.0134). At this late stage, representative images clearly illustrated the marked neuronal loss in these subfields compared to WT controls (Fig. 3C). Consequently, the overall neuronal density across the entire HPC was significantly reduced in 18-month-old 3xTg-AD mice (Fig. 3D).
Astrocytes in the HPC also underwent progressive morphological changes with age (Fig. 3E and S4C). By 18 months of age, astrocytes in both WT and 3xTg-AD mice showed significant morphological remodeling, characterized by enlarged cell bodies and thickened processes, compared with their 12-month-old counterparts (Fig. 3F and S4E, WT: 383.5 µm3 vs. 463.3 µm3, P = 0.0016; 3xTg-AD: 335.6 µm3 vs. 418.0 µm3, P = 0.0011). The density of astrocytes in the HPC remained stable across ages and genotypes (Fig. 3G and S4D). However, microglia in the HPC exhibited earlier activation than astrocytes. By 7 months, 3xTg-AD mice showed a significant increase in microglial reactivity, with a higher proportion of cells adopting a reactive morphology (Figs. 3I and 30.1% in 3xTg-AD vs. 56.5% in WT; P = 0.0038). This activation became widespread by 18 months (Fig. 3I). In 3xTg-AD mice with established pathology, both astrocytes and microglia were frequently found in close proximity to Aβ plaques and pTau tangles (Fig. 3K). Quantitative analysis confirmed this observation but revealed a difference from the Ent: from the initial emergence of detectable aggregates at 7 months, both glial cell types were closely associated with the pathology, and no significant difference was observed in the percentage of aggregates associated with either cell type at any age point examined (Fig. 3L). This suggests that, in contrast to the Ent, the hippocampal environment fosters the active engagement of both astrocytes and microglia with pathological aggregates from the early stages.
Taken together, 3xTg-AD mice exhibit severe deficits in hippocampal neurogenesis and early hippocampal microglial activation. These alterations provide insights into the unique vulnerability of the HPC in AD pathology and identify potential cellular targets for reliable interventions.
CCK Presents the Earliest and Region-Specific Downregulation in 3xTg-AD Mice
CCK, a neuropeptide involved in synaptic plasticity and memory formation (Li et al. 2014; Chen et al. 2013, 2019; Zhang et al. 2005; Feng et al. 2021) shows reduced levels in brain regions vulnerable to AD, suggesting its potential as an early biomarker of cognitive decline (Zhang et al. 2024; Plagman et al. 2019). To explore the impact of AD pathology on CCK expression in the brain, we quantified mRNA levels of CCK and its receptor CCK-BR across different ages and brain regions in 3xTg-AD and WT mice. For comparison, we simultaneously assessed the expression of other key genes associated with synaptic and neuronal function, including brain-derived neurotrophic factor (BDNF), glutamate decarboxylase 1 (GAD1), solute carrier family 1 member 2 (SLC1A2), and synapsin I (SYN1), which are known to decline with age and in AD (Mirza and Zahid 2018; Gao et al. 2022; Hill and Gammie 2022).
At 7 months of age, expression levels of BDNF, GAD1, SLC1A2, and SYN1 remained comparable between 3xTg-AD and WT mice across all regions examined (Fig. 4A-F). Notably, CCK was significantly downregulated specifically in the Ent of 3xTg-AD mice (Fig. 4A, 0.803 in 3xTg-AD vs. 1.000 in WT; P = 0.0421), whereas no such change was observed in the HPC or other cortical regions (Fig. 4B-F), suggesting the specific earliest vulnerability of CCK in the Ent. In the earlier stage of AD (2–5 months of age), expression levels of CCK and CCK-BR in the Ent of 3xTg-AD mice were comparable to those in age-matched WT mice (Figure S5).
By 12 months, as the disease progressed, significant downregulation of CCK and CCK-BR extended to multiple brain regions in 3xTg-AD mice, accompanied by marked reductions in BDNF, GAD1, SLC1A2, and SYN1 expression (Fig. 4A-F). By 18 months, these degenerative trends became more pronounced, with CCK exhibiting the most pronounced reductions across all brain regions analyzed (Fig. 4A-F).
Together, these results suggest that CCK expression is selectively and earliest impaired in the Ent during AD progression, supporting its potential role as a regional-specific biomarker for early AD detection.
CCK-4 Rescues Cognitive Function, Motor Learning, and Synaptic Function Across Disease Stages in 3xTg-AD Mice
To evaluate the therapeutic potential of CCK in AD, we first longitudinally characterized the progression of cognitive, motor, and synaptic deficits in 3xTg-AD mice from 2 to 18 months of age. The results of this characterization (presented in Fig. 5B, F, G and Figure S7) defined the specific ages for subsequent therapeutic intervention. Based on the established onset of significant deficits, separate groups of 3xTg-AD mice at these defined ages received acute administration of the CCK-BR agonist CCK-4 or vehicle (VEH) prior to behavioral training or electrophysiological recording.
We used the novel object recognition (NOR) test to assess cognitive function and the rotarod test to evaluate motor learning in mice (Fig. 5). During NOR training, all groups showed no inherent preference for object location (Fig. 5B, D), confirming unbiased exploration. In the test phase, however, 7- to 18-month-old 3xTg-AD mice exhibited significantly lower recognition indices (RI) than age-matched WT controls, indicating progressive impairment in recognition memory (Fig. 5B). Acute CCK-4 administration significantly improved NOR performance in 3xTg-AD mice across all ages tested, as shown by higher RI values than those in VEH-treated controls (Figs. 5D and 7 months: CCK-4 68.1% vs. VEH 60.9%, P = 0.0052; 12 months: 60.4% vs. 53.4%, P = 0.0065; 18 months: 58.1% vs. 50.9%, P = 0.0047). Loss of motivation or apathy often accompanies memory impairment in AD (Förstl and Kurz 1999). Consistent with this, 3xTg-AD mice spent less time exploring objects than WT mice from 7 months of age (Figure S6B, C), a motivational deficit not rescued by CCK-4 (Figure S6E, F). Sex-based analysis revealed no significant differences between males and females in NOR performance (Figure S6A-F).
In the rotarod test, 3xTg-AD mice performed similarly to WT controls at 2–7 months of age, but showed significantly shorter latencies to fall at 12 and 18 months, indicating impaired motor coordination and learning in later disease stages (Fig. 5F-G). Notably, acute CCK-4 treatment significantly improved motor learning performance in older 3xTg-AD mice, evident in steeper learning curves and longer fall latencies on Day 4 and Day 5 (Figs. 5I and 12 months: Day 4 CCK-4 256 s vs. VEH 208 s, P = 0.0157; Day 5 CCK-4 277 s vs. VEH 219 s, P = 0.0014. 18 months: Day 4 CCK-4 148 s vs. VEH 113 s, P = 0.0116; Day 5 CCK-4 159 s vs. VEH 123 s, P = 0.0172). Sex-grouped analysis showed no difference between males and females in the rotarod test (Figure S6G-H).
To assess synaptic function across different disease stages, we investigated LTP, a cellular correlate of learning and memory, in cortical and hippocampal slices using ex vivo electrophysiology. We focused on these regions as they are primary targets of entorhinal CCK projections, allowing us to evaluate the functional impact of entorhinal pathology on downstream memory circuits. After obtaining a stable baseline of field excitatory postsynaptic potential (fEPSP), LTP was induced by theta-burst stimulation (TBS). At 2 months of age, 3xTg-AD mice showed similar TBS-induced LTP in both the cortex and HPC compared to age-matched WT mice, indicating intact synaptic plasticity at this pre-symptomatic stage (Figure S7A, C). However, significant impairments in LTP magnitude emerged in 3xTg-AD mice from 5 months of age, became pronounced at 7 months, and progressively worsened at 12 and 18 months (Figure S7B, D), reflecting the progression of cognitive deficits. We next asked whether pharmacological activation of the CCK-BR could rescue these synaptic impairments. Remarkably, infusion application of CCK-4 during recording effectively restored synaptic plasticity in 3xTg-AD mice from 7 to 18 months of age (Fig. 6A-D). The rescue effect was evident shortly after TBS and was sustained throughout the one-hour recording period. Quantitative analysis of the normalized fEPSP amplitude averaged over the final 15 min confirmed the robust effect of CCK-4 (Fig. 6E, F). Relative to the pre-TBS baseline, cortical LTP in CCK-4-treated slices reached 134.9%, 130.6%, and 118.0% at 7, 12, and 18 months, respectively, values that were significantly higher than those in VEH-treated slices (Fig. 6E). Similarly, in the HPC, CCK-4 treatment potently enhanced LTP to 163.2%, 154.9%, and 136.1% of baseline at the same ages (Fig. 6F). These results demonstrate that acute CCK-4 treatment can significantly ameliorate deficits in synaptic plasticity across mild to severe stages of the disease in both important brain regions.
In summary, acute CCK-4 treatment significantly improved cognitive performance, motor learning, and synaptic plasticity in 3xTg-AD mice across mild to severe disease stages, demonstrating the remarkable therapeutic effects of CCK-BR agonist on AD.
HT-267, an Optimized CCK-4 Analogue, Is a Promising Drug Candidate for AD
Given the selective downregulation of CCK in the Ent during early AD, we next explored whether long-term CCK treatment could mitigate disease progression. We conducted a 90-day preventive treatment study in a group of 6-month-old 3xTg-AD mice (n = 20), a stage preceding overt cognitive and synaptic dysfunction (Fig. 5B, G and Figure S7E-F), randomly assigned to receive daily intraperitoneal injections of either HT-267 (n = 10) or VEH (n = 10) (Fig. 7A). HT-267 is an optimized CCK-BR agonist with a prolonged half-life, which we characterized in the present study. Throughout the treatment period, biweekly OFT revealed no significant differences in locomotor activity or motivational behavior between the HT-267 and VEH groups, indicating the absence of adverse behavioral effects from long-term HT-267 administration (Fig. 7B, C).
The NOR test was conducted for all mice before and after the 90-day treatment to assess cognitive changes. Although both groups showed comparable baseline performance (pre-treatment RI: HT-267 vs. VEH, P > 0.05), HT-267-treated mice exhibited a significantly higher RI than VEH-treated controls after treatment (61.04% vs. 53.32%, P = 0.0109) (Fig. 7D), demonstrating that HT-267 treatment attenuated age-related cognitive decline. Following the final behavioral assessment, animals from each group were allocated to terminal procedures: six mice per group for ex vivo electrophysiology and the remaining four for immunohistochemistry (IHC).
To evaluate synaptic plasticity, we conducted ex vivo electrophysiological recordings on hippocampal and cortical slices. Long-term HT-267 treatment resulted in significantly enhanced LTP compared to VEH controls (Fig. 7E-F; Cortex: HT-267 118.3% vs. VEH 111.3%, P = 0.0021; HPC: HT-267 132.3% vs. VEH 126.0%, P = 0.0171). We further assessed synaptic structural integrity by quantifying the expression of postsynaptic density protein 95 (PSD-95), a key scaffolding protein essential for maintaining excitatory synapses (Keith and El-Husseini 2008; Chen et al. 2011; Sturgill et al. 2009). In the HPC, HT-267 treatment increased dendritic PSD-95 immunolabeling by 25.7% compared to VEH controls (Fig. 7G-H; P = 0.0018). Consistently, the density of PSD-95-positive neurons was significantly higher in the cortex of HT-267-treated mice (Fig. 7I-J; HT-267 1042 cells/mm3 vs. VEH 895 cells/mm3, P = 0.0376), suggesting improved synaptic connectivity.
Taken together, long-term treatment of HT-267 improved memory function, enhanced synaptic plasticity, and preserved synaptic structure in 3xTg-AD mice. These results support the potential of CCK-BR agonists as disease-modifying therapies for AD, capable of delaying cognitive decline and strengthening neural circuitry integrity.




Comments (0)