
Remember me

Recent advances in the understanding of anti-tumor immunity have propelled immunotherapy as an effective treatment option against a wide variety of cancers (1). To date, most of the approved cancer immunotherapy modalities are monoclonal antibodies (e.g. checkpoint inhibitors and T-cell bispecific antibodies), recombinant cytokines (e.g. aldesleukin, multiferon, and Anktiva), or cancer vaccines that are designed to promote anti-tumor immunity (2). In recent years, immunotherapies that utilize cells as treatment modality have grown in popularity (3). These cellular immunotherapies are collectively called adoptive cell therapies (ACTs), since they involve the adoptive transfer of immune cells isolated from patient’s own blood (autologous cell therapy) or a donor’s blood (allogeneic cell therapy) following genetic editing (4, 5). ACTs have demonstrated superior and persistent anti-tumor responses compared to conventional therapies. The potent efficacy elicited by ACT is attributed to the ability of immune cells to initiate a cascade of multi-faceted events that are designed to either directly kill cancer cells or render the tumor microenvironment inhospitable to cancer cells. Furthermore, the self-renewal capability of ACT means that a persistent response of up to a few years, or even a decade, could be possible with a single dose of the immune cell products (3, 4, 6, 7).
The past four decades of research has resulted in the development of a series of ACTs, including tumor infiltrating lymphocyte (TIL) therapy, chimeric antigen receptor (CAR) T cell therapy, T cell receptor (TCR) engineered T cell therapy, CAR natural killer (NK) cell therapy, and CAR macrophage therapy (7). TIL therapy products consist of a pool of lymphocytes collected from a patient’s tumor. This pool of lymphocytes contains a mixture of T cells with different phenotypes that target a wide range of tumor antigens (8). CAR-T and TCR-T cell therapies are engineered T cell therapies with relatively defined characteristics that target a specific antigen expressed by cancer cells. CAR-T cells utilize a CAR construct, which is a genetically engineered receptor that contains an extracellular antibody fragment that binds to the tumor antigen and an intracellular signaling domain that activates T cells (9). In contrast, TCR-T cells are genetically altered to express a TCR construct that targets a specific tumor antigen. These TCR constructs can be based on naturally occurring TCR with high specificity against a tumor-associated antigen, or an engineered TCR that targets a neo-antigen that is specifically expressed in cancer cells (10). Beyond T cells, scientists have also designed NK cells and macrophages that carry CAR. Although CAR-NK cell and CAR-macrophage therapies have recently received widespread interests, T cell therapies are clinically proven in cancer treatment, and this review will focus on adoptive T cell therapy (TCT).
TCTs have been investigated in multiple indications, ranging from hematopoietic cancers, such as lymphomas and myelomas, to a broad spectrum of solid tumors (Table 1 and Supplementary Table 1). CAR-T cell therapy has enjoyed significant success thus far for treatment of blood cancers (11). Indeed, all except two currently approved TCTs are CAR-T cells targeting blood cancers such as B-cell acute lymphoblastic leukemia (B-ALL), multiple myeloma (MM), or B-cell non-Hodgkin lymphoma (NHL) (12). In contrast, treatment of solid tumors using either CAR-T cells or TCR-T cells have shown limited clinical success. One of many factors that may potentially contribute to this is densely packed solid tumor space that hinders tumoral penetration of TCT products (7). Moreover, since T cells are highly plastic and heterogenous, patient responses to TCTs are often variable. Finally, unlike traditional antibody-based immunotherapy, TCT products are “living” drugs, as they have the potential to expand in the patients. The self-replicating nature of the TCT is ideal for anti-tumor efficacy, but it poses challenges for managing toxicity, since treatment disruption is not straightforward (13). Furthermore, due to this self-replicating nature of TCTs, the traditional pharmacokinetics principles, such as dose-exposure relationships, do not often apply to TCTs.
Table 1 Summary of Doses and Treatment Outcomes of Adoptive T Cell Therapy in Clinical TrialsCurrent shortcomings of TCTs can be overcome by a better understanding of the cellular kinetics in whole blood (CK) and biodistribution of TCTs. For instance, it has been shown that anti-tumor effects of CAR-T cells or TCR-T cells are correlated with the expansion and persistence of the TCT products in patients (14, 15). Hence, an integrated understanding of the factors influencing the CK (e.g. donor specific factors, cell phenotype, engineering methods) and strategies to prolong T cell persistence could improve TCT efficacy. Fully characterizing the interpatient variability in TCT CK could help us understand the heterogeneity of patient responses (16). A detailed insight into what controls the penetration of TCT products into the tumor site could also help us better design TCT against solid tumors (17). Finally, strategies to minimize toxicity and maximize efficacy could be developed from our understanding of how cell proliferation affects the TCT CK (18). In this review paper, we will discuss current understandings of the TCT CK and biodistribution, focusing on the underlying biological factors that control them, and their effects on pharmacological responses of TCTs. We will also provide an overview on the in vivo, in vitro, and in silico models to assess TCT CK and biodistribution, as well as a brief discussion of bioanalytical methods to evaluate TCT CK and biodistribution.
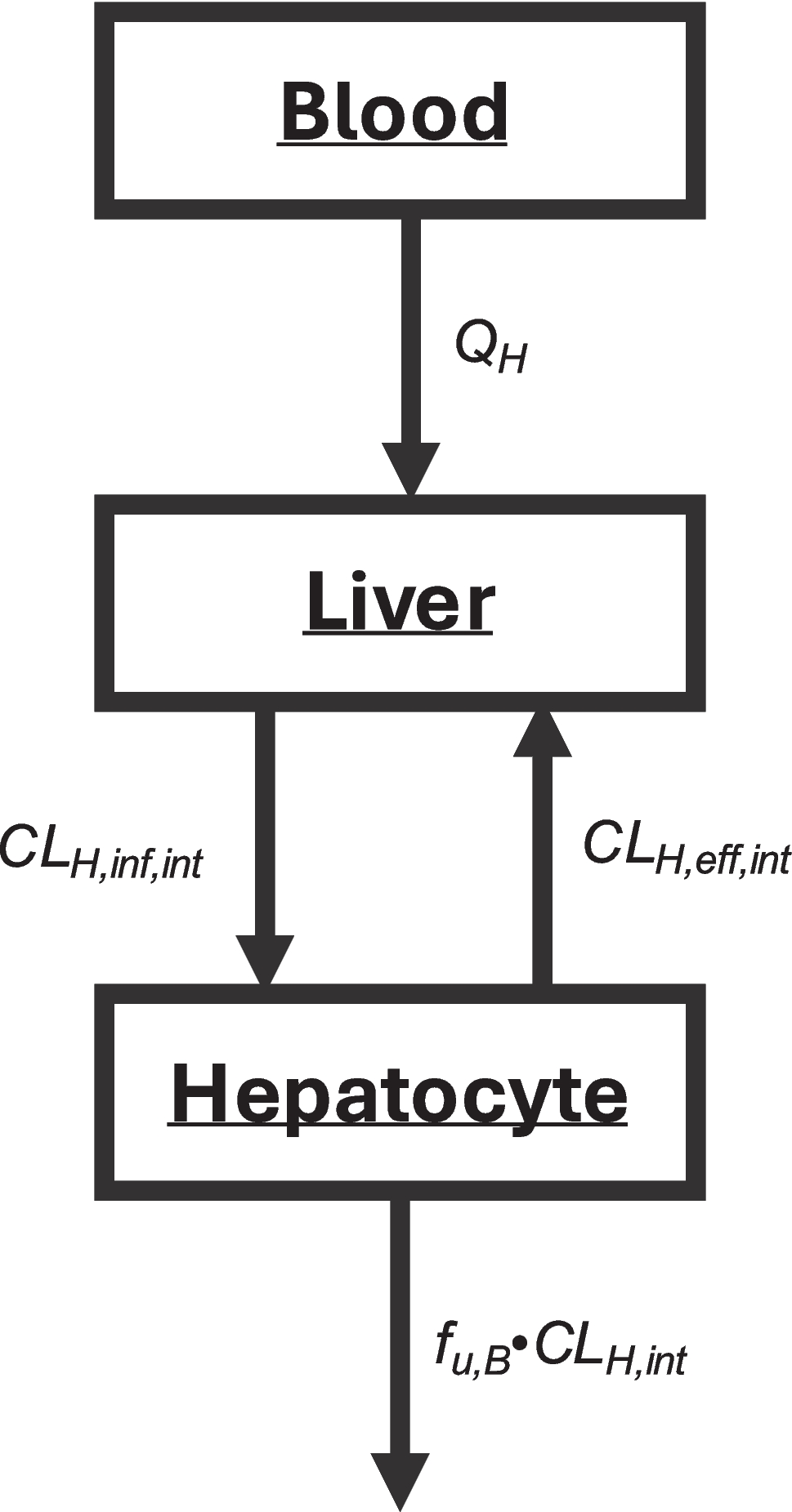
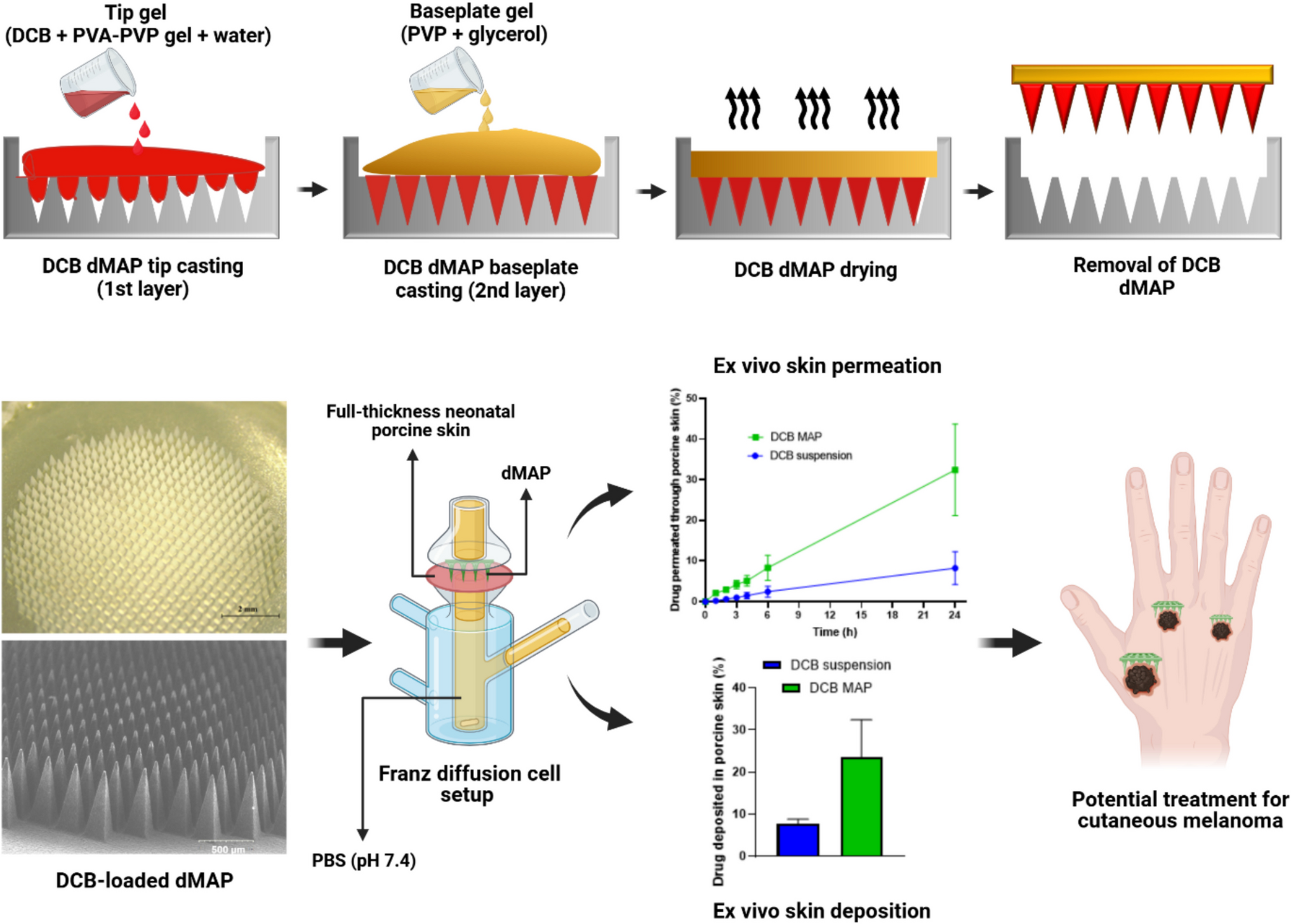
Overview of Adoptive T Cell TherapyTCTs can be broadly characterized into three formats: TIL therapy (19, 20), CAR-T cell therapy (21), and TCR-T cell therapy (22, 23). The production of TCT is a multi-step process that includes isolation of T cells from a patient’s or donor’s blood, activation of T cells for ex-vivo survival, gene-editing to express CAR or TCR, ex-vivo expansion of T cells (to enable desired dose of cell product), and intravenous infusion of T cell products into cancer patients. TCT products that are introduced into the patients are usually engineered with receptors, as well as costimulatory domains and cytokines, that help these cells in tumoral recognition and survival (Fig. 1a) (24). Since CK of TCT is controlled by the underlying biological properties of the TCT product (25), we will detail in this section the biological design and production process of TCT products.
Fig. 1
Adoptive T cell therapy (TCT). a Workflow for manufacturing and administering adoptive TCT. The steps in this workflow include isolation, activation, genetic engineering, expansion, and infusion of the patient-derived T cells. Factors relating to manufacturing process, patient conditions, and the design of TCT products that affect CK, biodistribution, and efficacy of TCT are listed. Autologous TCT are driven from T cells collected from patients’ own bodies. In contrast, allogeneic TCT are driven from T cells isolated from donors. b-d Common types of adoptive T cell therapy include tumor-infiltrating lymphocyte (TIL) therapy (b), chimeric antigen receptor (CAR) T cell therapy (c), and T cell receptor (TCR) engineered T cell therapy (d). The key differences and target profiles for these therapies are summarized
Tumor Infiltrating Lymphocyte (TIL) TherapyThe TCT products used in TIL therapy are naturally occurring tumor infiltrating lymphocytes isolated from patient biopsy (26, 27). Unlike CAR-T cells and TCR-T cells, TILs are not genetically engineered. The first attempt at TIL therapy was performed by Rosenberg et al., who successfully demonstrated that TILs isolated from mouse tumors can be used to limit the growth of metastases in mice (19, 20). Following this successful demonstration in animal models, clinical investigation of TIL therapy resulted in varying success (28, 29). TILs express naturally occurring TCRs that recognize tumor antigens presented by major histocompatibility complex (MHC) on cancer cells (Fig. 1b). Since TILs are essentially natural products with heterogeneous TCR expression and antitumor properties, there are large inter-patient and inter-indication variabilities in the performance of TIL therapies. TIL therapies in patients with cervical cancer, breast cancer, and melanoma have resulted in objective response rates as high as 50% (28, 30, 31), and the FDA has recently approved Lifileucel as the first ever TIL therapy for the treatment of melanoma. However, TIL therapies for renal cell cancer and non-small cell lung cancer have shown limited success.
CAR-T Cell TherapyIn contrast to TIL therapies, TCTs using engineered T cells have enjoyed greater commercial success. These engineered T cells express a neo-receptor that targets a specific antigen expressed by cancer cells, thus making these cellular products less heterogeneous compared to TILs (32). Two types of engineered T cell therapy currently exist: CAR-T cell therapy and TCR-T cell therapy (5). CAR-T cell therapy is the more advanced of the two, with seven FDA-approved products in the US and one approval in the EU. Central to CAR-T cells’ tumoricidal ability are the engineered CARs, which are recombinant proteins comprised of an extracellular single-chain variable antibody fragment (scFv) or a variable heavy domain of heavy chain (VHH), a hydrophobic alpha helix transmembrane domain, and an intracellular domain composed of immunoreceptor tyrosine-based activation motifs (ITAMs) of T cell co-receptor CD3ζ. The extracellular scFv or VHH domain is designed to bind specifically to a tumor-associated or tumor-specific antigen on the surface of the cancer cells (33, 34). Upon the extracellular domain-tumor antigen binding, the intracellular ITAM domain is phosphorylated which initiates the signaling cascade that activates the CAR-T cells for tumor killing (13, 33). Since scFv or VHH is utilized for target recognition, a major advantage of CAR is that its targets are not restricted by MHC presentation. This means that as long as a tumor antigen is expressed on the membrane of the cancer cell, a CAR-T cell can be developed to target it. However, this non-reliance on MHC presentation is also a double-edge sword, as intracellular tumor antigens cannot be targeted with CAR-T cells (Fig. 1c) (9).
The first generation CAR-T cells demonstrated potent in vitro cytotoxicity; however, their in vivo anti-tumor activity was limited, owing to inability of these modified T cells to replicate and expand in the patients (35). To solve this issue, second generation CAR-T cells were developed in which the intracellular signaling domain of a co-stimulatory molecule, such as CD28, 4-1BB and OX40, was inserted into the CAR construct (36, 37). This co-stimulatory signaling domain amplifies the activation signal in the T cell once the CAR is engaged with the tumor target, thus greatly promoting CAR-T cell proliferation (38). To further improve on this, researchers have designed third generation CAR-T cells that include two co-stimulatory domains and one ITAM domain (35, 39, 40). Fourth generation CAR-T cells incorporate an additional domain that would activate the nuclear factor of activated T cells (NFAT). Once the CAR binds to its target, NFAT is phosphorylated and translocates to the nucleus where it will induce the production of cytokines such as IL-2, IL-12, IL-18, and IL-15. These cytokines enhance the cytotoxicity of CAR-T cells and activate host immune cells in the tumors (41). The design of the fifth generation CAR-T cells further enhances the CAR-T cell proliferation by including STAT3/5 binding domain in the intracellular portion of the CAR construct. Hence, JAK-STAT pathway will be activated once the CAR is engaged, leading to a more sustained T cell activation, proliferation, and cytokine release (42).
Blood and bone marrow cancers are easily accessible by TCT products that are given as intravenous infusion to patients. This has led to remarkable clinical benefits with CAR-T cells in hematologic malignancies with overall response rates (ORRs) ranging from 50 ~ 95% (Table 1). For instance, CD19 CAR-T cell YESCARTA (axicabtagene ciloleucel) showed ORR of 83% in large B-cell lymphoma (43), 82% in refractory large B-cell lymphoma (44), and 94% in follicular lymphoma (45). BCMA CAR-T cell CARVYKTI (ciltacabtagene autoleucel) showed ORR of 97% against multiple myeloma (46). Currently, most approved CAR-T cell products target either CD19 or BCMA for hematological malignancies. CAR-T cells that target CD22, CD4, CD7, and CD38 in various blood cancer indications are under clinical investigation (NCT03620058, NCT03829540, NCT03690011, NCT03754764). In contrast, the response rates for CAR-T cell therapy targeting solid tumors have been less impressive thus far. The ORRs of EGFR CAR-T cells in non-small cell lung cancer (47) and glioblastoma (48) were 18% and 10%, respectively. In patients with neuroblastoma, CAR-T cell targeting GD2 elicited an ORR of 32 ~ 63% (49). A major obstacle for the successful implementation of CAR-T cell therapy in solid tumors is probably associated with inability of CAR-T cell to target intracellular tumor antigens. Unlike intracellular tumor antigens, which are more likely to be tumor specific antigens (TSA), surface receptors on cancer cells are usually tumor-associated antigen (TAA), meaning they are expressed in low levels on healthy tissues. Since TAAs are frequently expressed in critical organs, CAR-T cells targeting TAAs for solid tumor indications can often result in on-target/off-tumor toxicities that are difficult to mitigate (7, 9).
TCR-T Cell TherapyTCR-T cells are genetically engineered to express a neo-TCR that can bind to a specific TAA or TSA with high affinity (10). Unlike CAR, which is an artificial construct, the neo-TCR is often a naturally occurring receptor discovered from T cells isolated from cancer patients or healthy volunteer (50). Other sources of these TCRs include T cells collected from human HLA transgenic mice (51, 52) or human peripheral blood mononuclear cells (PBMCs) stimulated with tumor antigens (53, 54). Antigen-specific T cell enrichment (55, 56), TCR sequencing (57,58,59), and bioinformatic techniques (60) are currently used to identify TCRs with high affinity to target specific TAAs or TSAs. In some cases, directed evolution through phage display has been used to further improve TCR affinity (60, 61).
TCR is a heterodimer containing α and β subunits, and the sequence diversity of these two subunits allows TCR to recognize a wide variety of antigens. TCR can only bind to antigens presented by MHC molecules (62). When a TCR-T cell encounters a cancer cell, the extracellular component of TCR binds to tumor antigen-MHC complex and then interacts with CD3 to activate T cell for tumor killing. While the requirement for MHC presentation limits the target repertoire and patient population that could benefit from TCR-T cells, the ability of MHC to present both intracellular and cell surface antigens allow TCR-T cells to bind to a larger proportion of TAAs or TSAs compared to CAR-T cells (Fig. 1d) (63).
Clinical development of TCR-T cell therapy lags behind CAR-T cell therapy, as TCR discovery was difficult prior to the widespread use of sequencing technologies. Nonetheless, development in TCR-T cell therapy has recently enjoyed a period of boom, since TCR-T cell therapy holds promise as an effective treatment for solid tumors. This is because the majority of the TSA in solid tumor are intracellular antigens (7). These intracellular antigens can include oncoviral proteins (e.g. HPV E6/E7 protein) or endogenous proteins with oncogenic mutation (e.g. BRAFV600E) (22). TCR-T cells targeting HPV E7 have been tested in the clinic for treatment of metastatic HPV-associated epithelial cancers, and the reported ORR was 50% (64). TCR-T cells have also been designed to target cancer-testis antigens, which are TAAs that only express in germ cells outside of cancer cells. Clinical trials have been conducted with TCR-T cells targeting NY-ESO-1 (65), MAGE-A4 (66), and MAGE-A10 (
Comments (0)