We recently detailed 5 deficiencies of the ECC (6) as enumerated here:
(a)
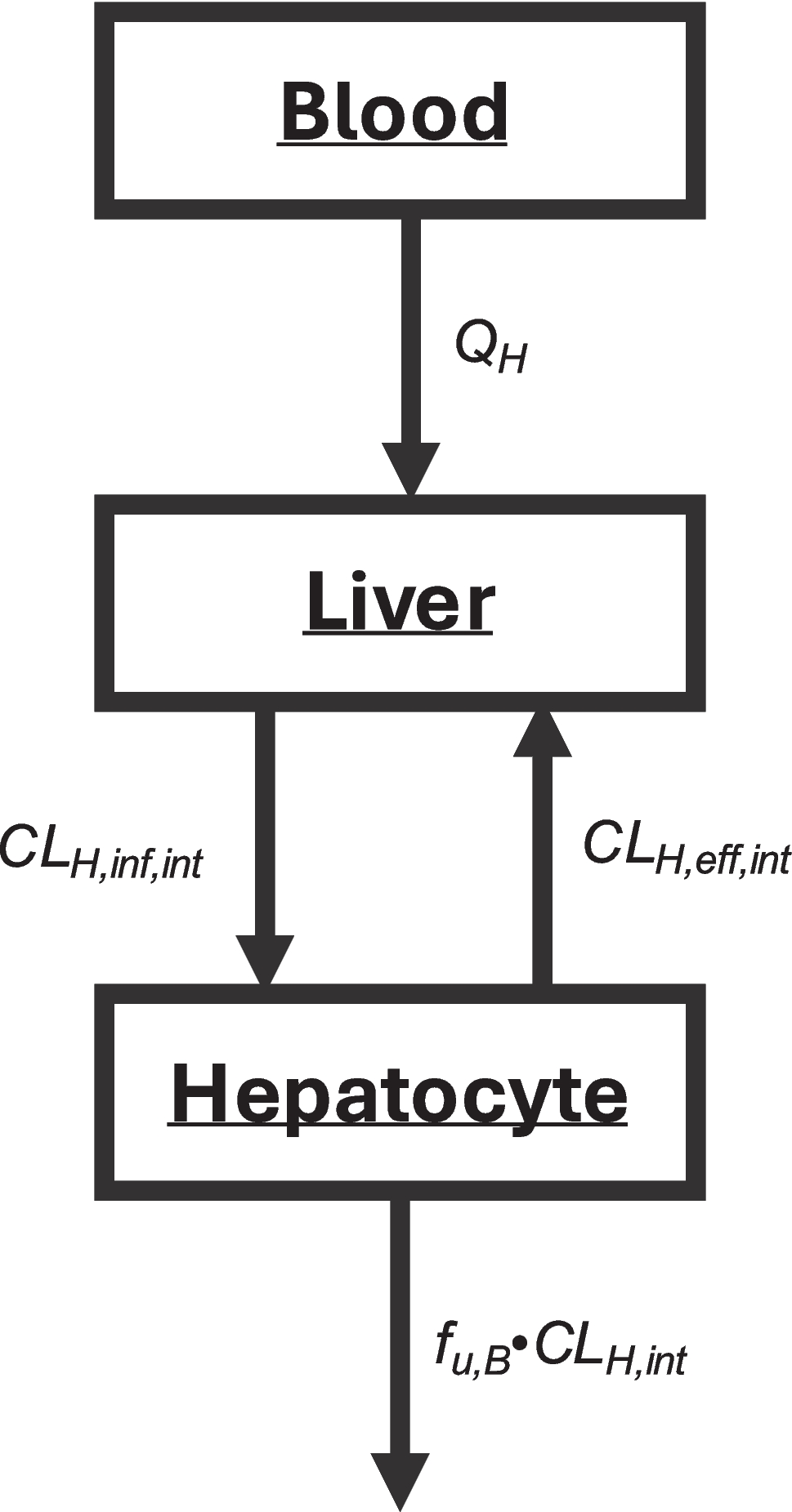
In Eq. 3, for hepatic basolateral transport (CLH,inf,int) to be rate-limiting, CLH,eff,int must be zero or negligible relative to CLH,int, such that CLH,int cancels out in the numerator and denominator. Then \(_=f_\cdot_\) and the “uptake process is important.” But why is it necessary for efflux transport to be zero or negligible compared to CLH,int for hepatocellular transport to be rate- limiting? Instead, if the net basolateral flux (i.e., the positive difference between influx and efflux) is much smaller than CLH,int, then \(_H=f_\cdot_-_)\). As discussed subsequently, this relationship is strongly influenced by how hepatobasolateral transport is quantified in vitro.
(b)
From Eq. 3, it is not possible for CLH,int to be rate-limiting unless it is assumed that basolateral influx and efflux are equal, and greater than CLH,int. That is, in the denominator CLH,int < < CLH,eff,int and then if CLinf,int = CLH,eff,int, \(_=f_\cdot_.\) However, if influx equals efflux, this represents a passive process, and the transporter clearances should not be included in the derivation. Instead, shouldn’t CLH,int be considered rate-limiting as long as it is significantly smaller than \(_-_)\)?
(c)
Eq. 3 was derived (3) based on the well-stirred model (WSM) hypothesis, where overall clearance within the liver is assumed to be independent of hepatic blood flow. However, this assumption is incorrect, as hepatic clearance is determined not only by the capacity of the liver to eliminate a drug, but also by the rate of delivery to the liver via blood flow. We have previously described the basis for this error in detail (2, 7).
(d)
Determining Kpuu, the ratio of the WSM-hypothesized average unbound liver concentration to the measured unbound systemic blood concentration, results in ECC values that are surprisingly always less than unity and lower than hepatic bioavailability FH (i.e., \(1-\frac_}_}\)), as we recently reported (8). We consider this is one of the key reasons to question the validity of the WSM and ECC and this finding remains unchallenged in the published literature.
(e)
When measuring only systemic concentrations to determine hepatic clearance, why wouldn’t hepatic organ clearance follow the same approach as kidney organ clearance, where, as shown in Eq. 1, the two membrane passage parameters are evaluated as a difference?
We subsequently examine the application of the ECC to renal clearance, as recently proposed by Asano et al. (9), highlighting both its key mechanistic limitations in quantitatively predicting renal clearance but also its practical utility in predicting magnitude of change in concentrations as a result of renal DDIs.
Adapting Kirchhoff’s Laws from Physics to Derive Clearance and Total Rate Constant Equations
In mid-2022, we discovered that it was possible to derive hepatic clearance equations independent of differential equations by adapting Kirchhoff’s Laws from physics (10), which we then expanded to derive all renal and hepatic clearance equations following both iv bolus and non-iv dosing routes, for both linear and nonlinear systems, as summarized in our 2025 Pharmacological Reviews paper (2) and presented in our even more recent tutorial simplifying the application and teaching of clinical pharmacokinetics (11). To summarize the approach, we recognized that determining clearance for both in vitro and in vivo pharmacological processes is based on the principle that when two or more rate-defining processes occur in parallel, the total value of the measured clearance equals the sum of those rate-defining processes. Conversely, when two or more rate-defining processes are in series, the inverse of the total measured clearance equals the sum of the inverse of those rate-defining processes. These relationships are given in Eqs. 4 and 5.
$$\frac1_}=\frac1_}+\frac1_}+....$$
(5)
A rate-defining process is one that, on its own, could potentially define a total clearance, one that is possible to measure experimentally when it solely determines the clearance, and for in series processes is a positive value (2, 11). A very simple general approach can be utilized to derive all clinically relevant clearance relationships, as given in Eq. 6.
$$\frac_}=\frac_}+\frac_}$$
(6)
Utilizing Eq. 6 to characterize the clinically relevant characteristics for hepatic and renal clearance, as well as for clearance measures when drugs are administered via routes other than iv bolus dosing, Eqs. 7 and 8 are the renal and hepatic clearance equations, respectively (2).
$$\frac_}=\frac_}+\frac_}+\frac_+_- _)}$$
(7)
$$\frac1_H}=\frac1_}+\frac1+\frac1\cdot_-_)}+\frac1\cdot_}$$
(8)
where QR is kidney blood flow and clearance from the absorption site (CLabs site) is a new term that we introduced (12) to describe the entering clearance of drug from the absorption site into the systemic circulation. For a first order absorption process, CLabs site may be simply considered as the product of the absorption rate constant (ka) multiplied by the volume of distribution of the drug at the absorption site (Vabs site). This volume term is the non-physiological parameter that defines the amount of drug at the absorption site divided by the concentration of drug at the absorption site. Vabs site has no more physiologic relevance than the volume of distribution steady state (Vss), but will certainly not be equal to Vss, as we previously described (12, 13). Any of the terms in Eqs. 7 and 8 can be the rate limiting in vivo clearance if it is markedly smaller than the other terms. For example, in vivo drug clearance may possibly be controlled by the dosage form manufacturer, independent of drug and patient characteristics, if in vivo CLabs site is the markedly smaller than all the other terms in Eqs. 7 and 8 (14).
We are frequently asked about the relevance of the difference in influx and efflux clearances in Eq. 8 for hepatic clearance if efflux is greater than influx. For the past 65 years, our field has had no difficulty in interpreting a negative clearance value for renal reabsorption clearance in Eq. 1 and it is known that for a number of drugs CLreab is greater than CLsec. That is, when the measured CLR is less than fuB · GFR. To answer this with respect to a potential negative basolateral transporter clearance, one must be aware of the methodology applied in adapting Kirchhoff’s Laws for clearance derivations and the definition of rate-defining process discussed extensively in our published manuscripts and as detailed above. Thus, it is theoretically possible for a negative renal transporter clearance to be a rate-defining processes as long as it is not greater than fuB · GFR, but in fact for such drugs the rate-defining process will always be the sum of the parallel (additive) transporter and filtration clearances since fuB · GFR will never be a large value that drops out of Eq. 1 when transporter effects are negative. For the liver, it is certainly possible that \(_\) can be greater than \(_\), but since the transporter clearance is in series with CLH,int, the negative value cannot be overcome and thus it is not possible for negative transporter clearances to be rate-defining, just as passive permeability clearances of drug passing back and forth between peripheral compartments cannot be rate-defining and thus do not appear on clearance equations. That is, as given in the definition of rate-defining processes, the process must be clinically measurable under some conditions. As we have emphasized in our publications, many processes can occur in vivo that are not rate-defining and thus do not appear in the clearance equations. In summary, while negative clearance values can occur and be physiologically meaningful in certain contexts like renal reabsorption, the structure of clearance pathways in the liver prevents negative transporter clearances from being rate-defining, ensuring that only clinically measurable, rate-defining processes appear in clearance equations.
There are two important aspects of transporter characterization and function that relate to Eqs. 7 and 8. The first is the relevance of considering organ blood flow. In the subsequent examples, we consider drug dosing via iv bolus, when CLabs site is infinite and therefore the first term in Eqs. 7 and 8 drop out. It could be argued that for most renally excreted drugs, renal blood flow will have minimal impact and need not be considered, since very few renally excreted drugs exhibit high clearance (15). However, intermediate clearance drugs can be affected by renal blood flow. For example (2), the measured CLR of metformin is approximately 600 ml/min in healthy young humans and with negligible plasma protein binding (fuB ≈ 1.0) and CLfiltration = GFR ≈ 120 ml/min. Therefore, previously the secretory clearance of metformin, \(_- _),\) was assumed to equal approximately 480 ml/min, as calculated by Eq. 1. However, when kidney blood flow is included (QR ≈ 1200 ml/min) as in Eq. 7, the secretory clearance is instead found to be 1080 ml/min, a value greater than twofold higher than estimated with the previous approach to estimating renal clearance. Thus, we have been potentially underestimating the contribution of tubular transport for drugs exhibiting moderate renal clearance values, such as metformin. This has significant implications when using in vitro measurements to predict in vivo parameters, i.e., IVIVE (in vitro – in vivo extrapolation). It is also highly relevant in predicting and defining the extent of transporter DDIs.
Just as we detailed the relationship between transporter activity and renal clearance above, the same holds for published analyses of hepatic clearance, hepatic transporter activity, and the effects of hepatic blood flow, as we reviewed (2). The extent of transporter interactions on fluvastatin with a hepatic blood clearance of 1134 mL/min, atorvastatin with a hepatic blood clearance of approximately 1140 mL/min, and repaglinide with a hepatic blood clearance of 877 mL/min, together with potentially other OATP substrates, will not be correct if hepatic blood flow is not considered (2).
The second important aspect is the recognition that transporter-enzyme interplay can only be unambiguously characterized following iv bolus dosing studies. For oral dosing, not only must the extent of first pass intestinal and liver metabolism be determined, but one must also determine the value of CLabs site, which is not the same as the intestine and liver metabolic clearance.
Limited Experimental Data and PBPK Analyses Support the Validity of the Extended Clearance Concept
Solving Eq. 8 for an iv bolus dose where QH is much larger than the two intrinsic clearance parameters results in Eq. 9
$$_H=\frac\cdot_\cdot(_-_)}_+(_-_)}=\frac\cdot_}_}_-_)}}=\frac\cdot(_-_)}_-_)}_}}$$
(9)
When \(\left(_-_\right)\gg _\), then \(_H=f_\cdot_\), and hepatic elimination rate-limits hepatic clearance. When \(\left(_-_\right)\ll _\), then \(_H=f_\cdot(CL}_-_)\), and hepatobasolateral transporters rate-limit hepatic clearance. For the latter case, using Sugiyama and Aoki (5) and Alluri et al. (16) as representative of the multitude of publications that conclude the rate-limitation of hepatic uptake based on the ECC is CLH,inf,int, it should be emphasized that none of these analyses can definitively prove that rate limiting uptake is only determined by the influx portion CLH,inf,int in the ECC Eq. 3, vs the net influx \(_-_)\) in Eq. 9. In vitro determination of these parameters will be addressed in the next section.
It is important to recognize that prior to our discovery of the ability to derive clearance and rate constant relationships using the adapted Kirchhoff’s Laws approach (10), the only methodology available was via differential equations, as initially proposed by Rowland et al. (17) where it is assumed that elimination within the liver is independent of QH. Therefore, this differential equation derivation of the ECC suffers from the five deficiencies detailed above. In contrast, Eq. 9 aligns more closely with both observed experimental data and known aspects of physiology, as it does not require CLH,eff,int to be negligible compared to CLH,inf,int for transporter effects to be rate-limiting, it explicitly defines how CLH,int can be the rate-limiting step for elimination even when transporter effects are present, it imposes no restrictions on the value of Kpuu, and it is consistent with kidney clearance models where transporter permeability is characterized as a net difference. While experimental data cannot definitively validate either the ECC or the adapted Kirchhoff’s Laws approach to determining CLH, the advantage of Eq. 9 is that it avoids the limitations inherent in the ECC and remains consistent with all available experimental evidence.
However, a challenge remains in applying the approach presented here to renal clearance IVIVE predictions. In clinical settings, the only measure possible in a patient is systemic drug concentration. As a result, it is difficult to directly translate individual in vitro transporter clearance measures into in vivo CLR estimates that account for the overall net clearance of \(_- _\). This is particularly true when multiple transporters are involved, such as bidirectional OCT2 at the basolateral kidney membrane and unidirectional MATEs at the brush-border membrane, as addressed by Asano et al. (9). Despite the mechanistic concerns we have outlined regarding the ECC, it is important to acknowledge that reasonable IVIVE predictions of renal transporter-mediated DDIs can still be achieved using both the approach presented here and the ECC and PBPK-based approaches, which will be of use to NME sponsors and the regulatory agencies, particularly in labeling recommendations. This apparent paradox can be explained by the nature of DDI predictions, which rely on detecting relative changes in systemic exposures rather than accurately quantifying absolute renal clearance. As a result, despite the deficiencies enumerated for the ECC approach, the ECC and PBPK models may provide sufficiently accurate predictions of DDI magnitude, even if they lack mechanistic precision. However, as we have emphasized, achieving a reasonable fit to experimental data with multiple components in the ECC (and even more components in PBPK models) does not constitute mechanistic validation. From a clinical perspective, particularly in the context of drug dosing adjustments in renal failure, we commend Asano et al. (9) for their valuable recognition that for OCT2/MATE substrates, the widely-adopted approach of adjusting drug dosing based on effective GFR remains appropriate, even in severe renal failure. This also holds true for OAT substrates in mild to moderate renal impairment. However, for OAT substrates in severe renal failure, CLR decreases on average by 60% relative to the effective GFR measurement. Importantly, there is significant variability observed in this metric among OAT substrates, with hydrochlorothiazide and famotidine decreasing by 95% and 96%, respectively, while adefovir exhibits a modest increase of 8%.
Sugiyama and Aoki (5) summarized the work of Ueda et al. (18) with respect to the effect of inhibitors on the apparent intrinsic clearance, indicating that when hepatic basolateral transport is the rate-limiting clearance parameter, Eq. 10 is the relevant relationship.
where CLH,all,int,DDI and CLH,all,int,control are, respectively, the overall intrinsic clearance values during a drug-drug interaction (DDI) and under control conditions, Iinf is the concentration of the influx transporter inhibitor and Ki,inf effects is the inhibitory constant of the inhibitor on the influx transporter protein. When in the ECC CLH,eff,int > > CLH,int, a product is proposed (5, 18) in the denominator to define maximum inhibition, as given in Eq. 11, where efflux transport or metabolism is the alternate elimination interaction.
$$_=\frac_}}})\cdot(1+\frac}})}$$
(11)
The authors (5, 18) note that “estimating unbound inhibitor concentration in the cell remains a challenge when applying this methodology”. Although the authors used these equations to fit the experimental data, several aspects of the analysis warrant further scrutiny. First, we agree that estimates of unbound inhibitor concentration within the cell based on in vitro measures are a challenge. Measures of hepatic clearance are multiplied by systemic concentrations to predict drug loss; therefore, it is only systemic concentrations of the inhibitor that can be determined and Ki estimates will be in terms of systemic concentrations. Second, the adapted Kirchhoff’s Laws approach (Eq. 9) leads to the correct estimate of the maximum hepatic inhibition as given in Eq. 12, rather than Eq. 11.
$$\frac1_H}=\frac1_}+\frac1+\frac}}}\cdot_-_)}+\frac}}}\cdot_}$$
(12)
In Vitro Determination of Transporter Clearances
Based on differential equation derivations, it is proposed that permeability clearances for hepatobasolateral transporter-mediated influx can be experimentally determined in the same way that is conducted for determining passive permeability values, i.e., measuring initial passage into the hepatocyte or the hepatocellular membrane so that concentrations driving efflux are minimal and can be ignored. Transporters function either to enhance the absorption and distribution of beneficial compounds, or to serve as a protective mechanism in limiting the entry of xenobiotics into the body and specific organs. Transporters would not adequately serve this latter function if they only effluxed drug from outside of the membrane. Thus, attempts to quantitatively determine directional transporter clearances based on initial rates of membrane passage are not valid. Measurements of initial-rates of transporter clearance, whether for influx and efflux, reflect the net difference between influx and efflux clearances, as transport across a membrane inherently involves a balance between opposing fluxes. Since concentrations within the membrane cannot be directly measured, it is not possible to separate the influx from efflux components. Thus, within the adapted Kirchhoff’s Laws approach to hepatobasolateral transport, it is possible that the CLH,eff,int value is negligible compared to CLH,inf,int, as proposed in the ECC, however, this cannot be determined experimentally. Thus, Eqs. 8 and 9 provide the appropriate values of hepatic clearance regardless of the relationship between CLH,eff,int and CLH,inf,int, since only systemic drug concentrations can be measured. The in vitro hepatic uptake measurements change over time, as efflux reflects both the intramembrane efflux and the efflux of increasing drug concentrations outside the membrane. The excellent work of Ménochet et al. (19, 20) predicting clearance for drugs that are substrates for hepatic basolateral transport and metabolism and/or renal elimination using in vitro measures highlights the difficulties in validating mechanistic models. The authors assume that the in vitro measures only reflect uptake minus passive efflux and that active efflux is negligible, consistent with the ECC, within a two-compartment in vitro system. However, the in vitro data could equally reflect measures of influx minus efflux as proposed in the adapted Kirchhoff’s Laws, leading to the same predictive outcome. The proposed methodology based on adapted Kirchhoff’s Laws offers a more fundamental, assumption-light framework that can clarify mechanistic interpretation and better accommodate complex clearance processes (such as simultaneous active influx and efflux). This can improve understanding, model robustness, and potentially guide experimental design in ways that the current models cannot.


Comments (0)