In this report, we present analysis of immunogenicity data from more than 7,000 study participants from CAS + IMD clinical trials conducted during the SARS-CoV-2 global pandemic. The CAS + IMD clinical drug development program included numerous studies across different indications, administration routes, doses, and regimens. In addition to general observations about the humoral immune response to CAS or IMD, this large and complex dataset provided a unique opportunity to investigate the impact of various factors, such as sample collection timing, frequency of drug administration, dose of drug, and route of drug administration, on immunogenicity.
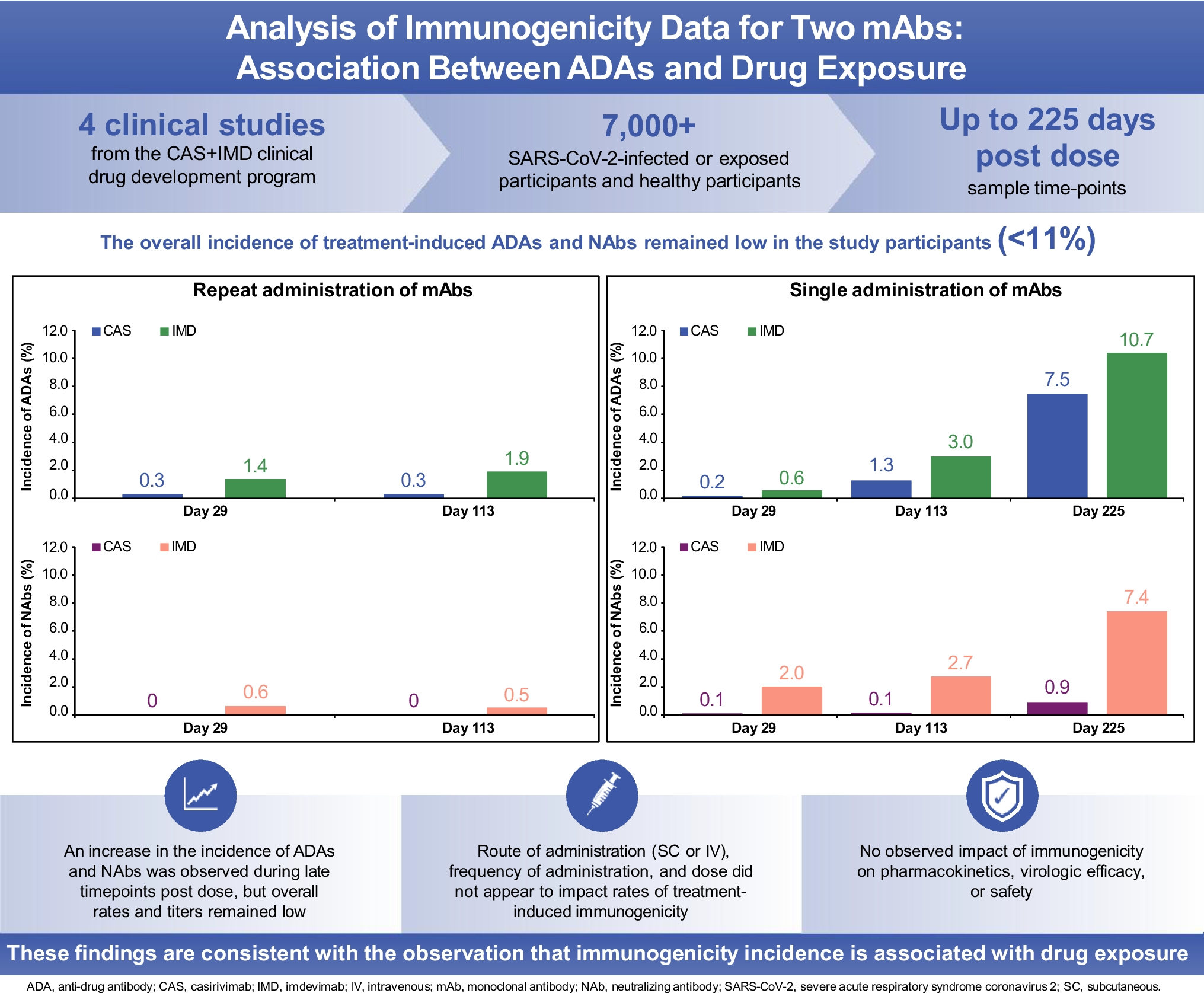
In the studies described here, we followed participants after drug administration for a longer duration than in typical clinical trials, where the last ADA timepoint is often only several months after the last dose. In some of our studies, immunogenicity was measured as late as 225 days post-last dose. While the rates and magnitude of treatment-boosted + treatment-emergent ADAs and NAbs for CAS and IMD remained generally low, the immunogenicity incidence increased over time after the last dose of CAS + IMD. This observed increase at late timepoints was unexpected as we have extensive experience with human mAbs using the same platform for protein engineering and manufacturing, where immunogenicity incidence is very low; however, we have not monitored ADA development as late as 225 days post-last dose in other clinical drug development programs. For protein biotherapeutics, the increased detection of ADA at late timepoints when drug concentrations declined, could potentially be due to interference of drug in the ADA assay at timepoints closer to administration when drug concentrations are higher. Importantly, the data described here for CAS + IMD does not support this hypothetical scenario as these assays were sensitive and drug tolerant, even at an early time post-dose (i.e., D29). In addition, at timepoints from ~ 4–8 months after administration (i.e., D113 and D225), drug levels were more than 2 orders of magnitude lower than the assay DTL, when potential for drug interference in the ADA assay is negligible (Supplemental Fig. 1).
This suggests that drug exposure levels may be associated with the development of ADA and maintaining a sustained high drug concentration in serum, via repeated drug administration, leads to reduced immunogenicity due to immune tolerance; using repeated and high-dose drug administration is a well-documented practice for some therapeutic proteins where obtaining immune tolerance is critical for treatment (14). Once drug administration is stopped, drug concentrations fall, and immunogenicity incidence increases with elapsed time; the elevated ADA incidence may be the outcome of affinity maturation of B-cell responses observable over the unusually long observation time in these studies, which results with ADA detection in the assays at late timepoints. These two scenarios are illustrated with Fig. 1 (i.e., low ADA incidence during repeated drug administration) and Fig. 2 (i.e., increasing ADA incidence with elapsed time after drug administration). Published data indicate that high concentrations of protein therapeutics may be tolerogenic, and conversely, lower exposures can result in increased probability of developing ADAs (14, 23). Although ADA incidences and titers to CAS and IMD were overall low, the gradual increase in ADA incidences over time and as concentrations of mAbs in serum correspondingly decrease are consistent with the hypothesis that lower systemic concentrations are generally more immunogenic.
The presence of drug in a serum sample may interfere in the detection of ADA. However, the ADA assays for both CAS and IMD were highly drug tolerant and were able to detect very low levels of ADA (~ 100 ng/mL) in the presence of drug concentrations approximately two orders of magnitude greater than the drug concentrations at early ADA collection times. Furthermore, in the single dose study (Study 2069), the ADA incidence increased from D113 to D225 when drug levels were already several orders of magnitude lower than the drug tolerance of the ADA assay. Therefore, the observed increase in ADA incidence at later timepoints is unlikely to be an artifact of decreasing levels of interference in the assays as drug concentrations decline.
There are minor differences in immunogenicity incidence between the two mAbs, with a slightly higher rate observed with IMD than with CAS. However, overall both mAbs had low ADA development, and any small distinctions may be the result of subtle differences between the separate assays for each drug (e.g., cut points, assay sensitivity etc.).
Irrespective of the population, at an early timepoint of D29, a low frequency of immunogenicity was observed. These data indicate that in a treatment setting, which is typically of short duration, there is a low risk for participants to develop ADA responses that impact efficacy. However, there is an increased risk of developing impactful immunogenicity for therapeutic proteins in prophylaxis indications when the drug is administered infrequently or when seasonal administration is required. Many types of at-risk participants are likely to be candidates for biotherapeutics, and not only those with a demonstrated inability to mount an immune response to vaccines (immunocompromised) that would otherwise need protection. Theoretically, the immunogenicity risk to the biotherapeutic in the disease prevention setting for immunocompromised participants may be mitigated by their own compromised immune system. The propensity for development of ADAs to increase after cessation of drug administration is therefore an important consideration in situations when protein therapeutics will be administered intermittently (24).
In addition to CAS and IMD being fully human mAbs, the fact that the drug target is exogenous may also contribute to the low observed immunogenicity. The SARS-CoV-2 virus has as a tropism for epithelial cells in the respiratory system and is normally absent in serum (or present at extremely low concentrations) (25). Therefore, the drug is found predominantly in an unbound state in blood. The lack of systemic target engagement in vivo and the absence of large molecular weight target:drug complexes may reduce the level of surveillance of CAS + IMD by the host immune system. Similarly, a lack of immunogenicity to mAbs with exogenous targets has been observed in other clinical trials for viral countermeasures (26, 27).
SC administration may be preferred over IV administration due to participant convenience, compliance and possible comfort. However, there is a perception that SC administration is more immunogenic than IV administration, although the evidence is not conclusive and seems to differ between therapeutic proteins (4, 28, 29). Importantly, we observed that the route of drug administration did not appear to significantly impact CAS or IMD immunogenicity incidence or titer. This is consistent with the observation that other factors (e.g., non-human sequences, intermittent dosing, adjuvants) may have greater impact on immunogenicity than route of administration (29).
Overall, the frequency and magnitude (titer) of immunogenicity to both mAbs was low. When immunogenicity was observed, it was more often detected at lower drug concentrations and at later study timepoints (i.e., more than 3 months after last dose). Importantly, we found that concentrations of CAS and IMD were similar between ADA-positive and ADA-negative participants, indicating immunogenicity did not impact PK. Administration of CAS + IMD resulted in a favorable safety profile, including being well-tolerated, across studies as previously reported (11, 12, 30, 31), and there were no identified clinically significant effects of anti-CAS and anti-IMD antibodies on safety and effectiveness over the treatment duration.


Comments (0)