
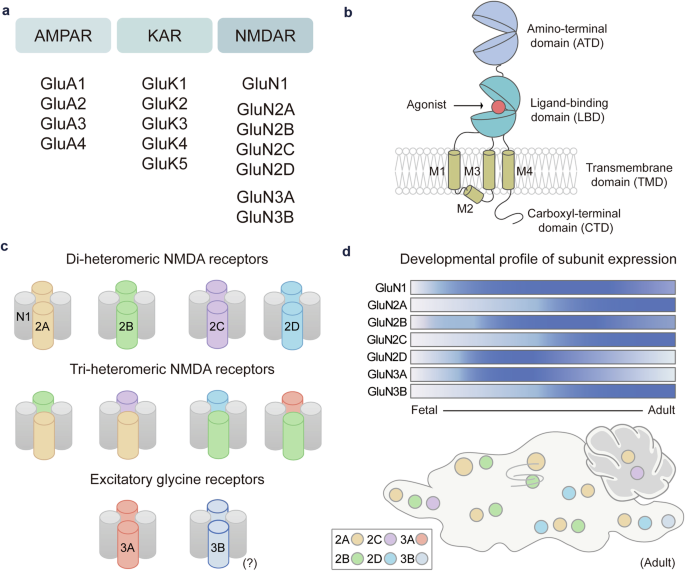
Remember me
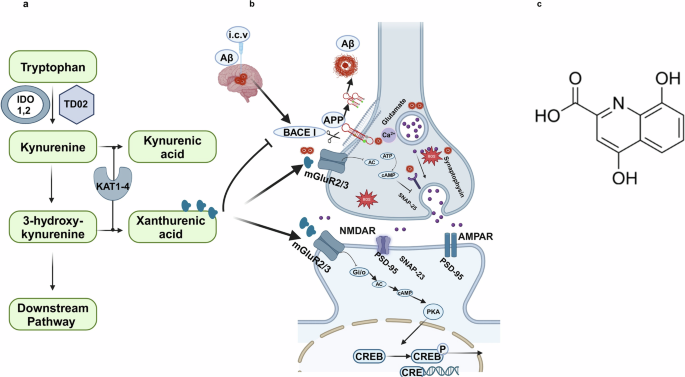
(XA; Sigma-Aldrich, USA, Cat. #D120804) and Aβ peptide (Aβ1–42; GenScript, Cat. #RP10017) were used in this study. Primary antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX, USA) and are listed in Supplementary Table S1.
AnimalsAdult male C57BL/6 N wild-type mice (8–10 weeks old, 25–30 g) were purchased from Samtako Bio (Osan, Republic of Korea). Mice were acclimated for 2 months under standard housing conditions (23–25 °C, 60% ± 10%, 12-h light/dark cycle) with ad libitum access to food and water. The sample size was determined based on a priori study, which investigated kynurenic acid (KA) [29]. To reduce bias and improve the rigor of our findings, randomization and blinding procedures were implemented throughout the experiment. These results were representative of three different tests. All animal experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Department of Biology, Division of Applied Life Sciences, Gyeongsang National University (Approval ID: GNU-230614-M0129-01).
Intracerebroventricular (i.c.v.) injection of Aβ1–42Aβ1–42 peptide was dissolved in sterile isotonic saline (1 mg/mL) and incubated at 37 °C for 4 days to promote oligomer formation. A 5 μL aliquot of this oligomerized solution was stereotaxically injected into the right lateral ventricle using a Hamilton microsyringe under ketamine (0.1 mL/kg) and xylazine (0.05 mL/100 g) anesthesia. Coordinates relative to bregma were: AP: −0.2 mm, DV: −2.4 mm, ML: 1.0 mm. The injection was administered over 5 min to minimize tissue damage.
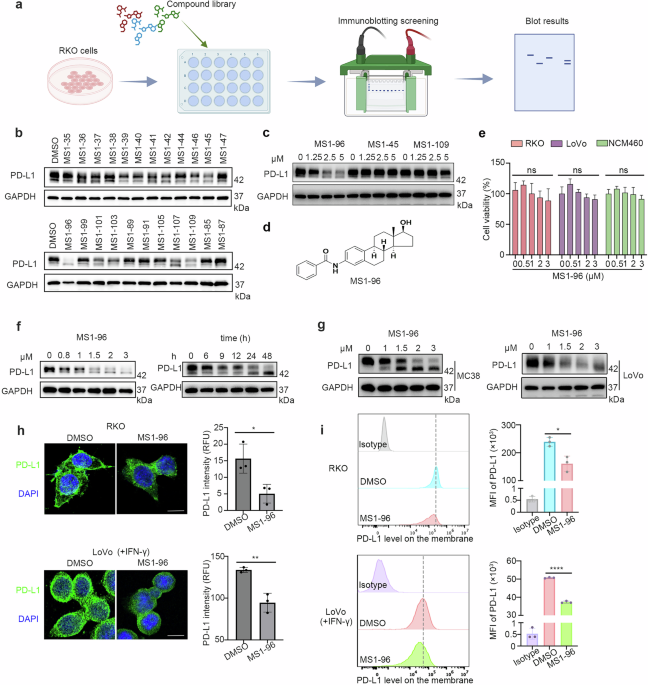
Intranasal administration of XATo ensure proper body positioning for effective awake i.n. delivery, the mice were acclimated to handling i.n. dosing approximately 3 weeks before treatment, as suggested by Hanson et al. [2]. Forty mice were randomly divided into four groups (n = 10 mice/group): Group 1 (Control) received 0.9% normal saline; Group II (Aβ induced) received Aβ1–42 (5 µL/5 min per mouse); Group III (XA treated) received Aβ1–42 followed by XA (0.5 μg/5 μL per nostril, daily for 6 weeks); Group IV (XA alone) received only XA identically to Group III without Aβ (Fig. 2). XA-alone group was included to assess cytotoxicity, oxidative stress, and neuroprotective outcomes. 100 mM XA was dissolved in dimethyl sulfoxide (DMSO) and diluted in cell culture media or saline to a final concentration of 0.1% DMSO. Since KA and XA are closely related metabolites with both exhibiting neuroprotective properties, and as no prior studies have assessed XA as a therapeutic intervention, the dosage was determined based on a previous study in male mice, which demonstrated that a dose of 0.5 μg significantly enhanced memory consolidation across various dosage regimens [30].
Fig. 2
Experimental design for investigating the therapeutic effects of intranasal XA administration in an Aβ1–42-induced AD mouse model.
Cognitive behavioral assessmentsNovel objective recognition (NOR)The novel objective recognition (NOR) test was conducted in a 90 cm × 60 cm × 35 cm open-field arena over three consecutive days: habituation (5 min, no objects), familiarization (10 min with two identical objects), and testing (10 min with one novel and one familiar object, 24 h after familiarization). Object interaction was scored by a blinded researcher. A discrimination index was calculated to quantify object recognition [3].
$$}}}\,\left(}}}\right):}}}=\, }}}-}}}/}}}\\ +}}}$$
$$}}}\,(}}}):}}}=(}}}/}}}+}}})\times 100$$
Y-maze testTo investigate spontaneous changes in behavior during exploratory activity, we utilized a Y-maze constructed from black-painted wood, featuring three arms measuring 50 cm in length, 10 cm in width, and 20 cm in height [4]. Briefly, after completing one trial, each mouse was put in the middle of the three arms and allowed to move freely about the Y-maze for 8 min. All arm entries and modifications were recorded. The percentage of spontaneous alternation (%) was calculated using the formula: (number of consecutive entries into three different arms/total arm entries − 2) × 100. A high percentage of spontaneous alternation indicated improved cognitive function, while a lower percentage suggested reduced cognitive performance.
Morris water maze (MWM)The Morris water maze (MWM) is typically regarded as an assessment of memory and spatial learning [5]. Following 14 days of mouse treatment, we performed the MWM test using the previously reported technique, with a few alterations [6]. The water maze device consists of a circular water tank (diameter: 100 cm, height: 40 cm) that is filled to a depth of 15.5 cm with water mixed with white ink and kept between 22 and 25 °C. In each of the tank’s quadrants, a 10-cm-tall, hidden transparent platform was positioned 1 cm below the water’s surface. Every mouse was given a starting point from the remaining three quadrants and given a 5-day trial period to locate the concealed platform. Mice were trained on the platform and kept there for 30 s if they could not investigate the hidden platform quadrant in 60 s. The amount of time spent in the target quadrant and the time it took to escape were measured for each experiment. A probe test was then performed to evaluate memory consolidation, and the process was carried out in the same way for every mouse, which involved removing the hidden platform. The number of crossings around the platform, the latency towards the platform, and the total amount of time each mouse spent in the target quadrant were recorded during the probe test. Video-based motion tracking software (SMART 3.0, Panlab Harvard Apparatus, Bioscience Company, Holliston, MA, USA) was used to record the data.
Cell culture and transfectionHuman neuroblastoma SH-SY5Y cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin, and maintained at 37 °C in a humidified incubator with 5% CO2. When cells reached approximately 70%–80% confluency, it was transfected with APPswe/ind gene cloned into the pCAX vector during Lipofectamine 3000 (Invitrogen, CA, USA) according to the instructions provided by the manufacturer. Cell viability, cytotoxicity, and caspase-3/7 activation (apoptosis) were all measured using the ApoTox-Glo Triplex Assay (Promega, WI, USA), which was carried out as previously described [7]. The GloMAX Multi Detection System (Promega, WI, USA) and a microplate spectrophotometer were used to measure the absorbance and fluorescence readings within a single 96-well assay. The XA dosage references (0, 3, 10, 30, and 100 μM) were selected based on a study that investigated KA [8].
Tissue collection and preparationAfter finalizing the cognitive behavior assessments, the mice were deeply anesthetized and euthanized. The animals were randomly selected for either quick decapitation and brain isolation, allowing for Western blot analysis or brain perfusion, for downstream immunofluorescence analysis according to previous protocols [9]. For Western blot analysis, the brain will be rapidly dissected (isolating the cortex, hippocampus, and olfactory bulbs), after which the brain samples will be snap-frozen using liquid nitrogen and stored at −80 °C. As directed by the provider (iNtRON Biotechnology, Inc., Sung-nam, Republic of Korea), the brain tissues were homogenized in a pro-prepTM protein extraction solution. After that, the samples were centrifuged for 25 min at 4 °C and 13,000 rpm. After collecting, the supernatants were kept at −80 °C. For tissue examination, mice (n = 5/group) were transcardially perfused with 4% ice-cold paraformaldehyde, and their brains were post-fixed in the paraformaldehyde for 72 h before being placed in 20% sucrose for the same amount of time. A CM 3050C cryostat (Leica, Germany) was used to cut 14-μm coronal slices from the brains after they had been frozen in the O.C.T. compound (A.O., USA). The sections were thawed and mounted on probe-on plus charged slides (Fisher, Rockford, IL, USA).
Western blot analysisWestern blots were carried out using the previously established methodology to assess the levels of many proteins associated with AD in the cortex, hippocampus, and olfactory bulb regions [10]. Bio-Rad protein assay kit (Bio-Rad Laboratories, CA, USA) was used to measure the amount of protein in tissues. An equivalent quantity of protein samples (25–30 µg) was used in the gel electrophoresis, which used 4%–12% BoltTM Mini Gels. Molecular weights were controlled using a broad-range pre-stained protein ladder (GangNam-StainTM, iNtRON Biotechnology). PVDF membranes were covered with gels containing various protein bands. To lessen the nonspecific protein binds, these PVDF membranes were next quenched for 1 h using skim milk (5% w/v skim milk in 1× TBST). The membranes were then treated with primary antibodies diluted at a ratio of 1:1000 dilutions for an entire night at 4 °C. Membranes were conjugated with appropriate secondary antibodies three times on the following day after being washed with 1× TBST for 10 min. Protein bands were identified using the ECL reagent (EzWestLumiOne, ATTO, Tokyo, Japan). The computer-based ImageJ program (v. 150, NIH Bethesda, MD, USA) was used to assess the protein expression levels on scanned X-ray films by densitometry analysis.
ImmunofluorescenceImmunofluorescent analysis was performed according to previous literature with little bit modifications [11]. Snap-frozen brains were used to create 14 µm coronal cryosections, which were then dried overnight and stained. Slides containing samples of cerebral tissue were blocked for 1 h using 5% normal goat serum after being cleaned twice for 10 min with PBS. The slides were then incubated at 4 °C for 24 h after being exposed to certain antibodies at a 1:100 dilution. The slides were stained with anti-mouse/-goat/-rabbit secondary antibodies for 2 h at a dose of 1:100 on the next day after being washed in PBS. After that, the process entailed 10 min of exposure to 4′,6-diamidino-2-phenylindole. A confocal laser microscope (FV3000 Operational Manual; Olympus, Tokyo, Japan) was used to observe the images after the slides were covered with coverslips and a mounting medium (DAKO). Confocal images were quantified, and relative integrated density (the sum of all the pixels within a particular region of the microscopic image and the mean grey value) was calculated using ImageJ software, and the graphs as well as statistical computations were carried out by using GraphPad Prism software (version 8.0, San Diego, CA, USA).
Antioxidant assaysGlutathione reduction was calculated using a previously developed approach by Ali et al. with minor adjustments [12]. 0.1 mL of tissue supernatant was combined with 2.4 mL of phosphate buffer stock solution and 0.5 mL of freshly made 5,5′-dithiobis (2-nitrobenzoic acid) stock solution to begin the activity. After 10 min, the intensity of the yellow color formed was measured at 412 nm using a spectrophotometer. The final results for GSH concentration were given in μmoles of GSH/g of material. Similarly, using the previously published Habig method, GST activity was measured at 340 nm using its typical substrate, 1-chloro-2,4-dinitrobenzene [13]. Catalase activity in intestinal tissue was measured and expressed as micromoles of H2O2 degraded per milligram of protein per minute, based on absorbance at 240 nm [14]. The impact of the XA therapy on SOD activity was examined. The SOD assay was performed as reported previously with little modifications. In short, the sample, pyrogallol (24 mM), and tris-EDTA (50 mM, pH 8.5) were combined in 96-well plates (200 μL reaction mixture), and the absorbance was measured at 450 nm. All activities were conducted in triplicate.
Additionally, using methods described by Utley et al., the lipid peroxidation (LPO) rate was determined by calculating the concentration of malondialdehyde (MDA) with small modifications [15]. 580 μL of 0.1 M phosphate buffer (PH 7.4), 200 μL of supernatant, 20 μL of mM ferric chloride, and 200 μL of 100 mM ascorbic acid make up the assay combination, which is then incubated for 60 min at 37 °C in a water bath. 1000 μL of 10% trichloroacetic acid and 1000 μL of 0.66% thiobarbituric acid (TBA) were added to the samples to terminate the reaction after 1 h of incubation. After 20 min in a water bath, the tubes were chilled in an ice bath and centrifuged for 10 min at 3000 × g. The concentration of thiobarbituric acid reactive substances (TBARS), which was represented as nM TBARS/min/mg protein, was measured at 535 nm using supernatant absorbance and a blank that contained all reagents except the test sample.
Statistical analysisImageJ software was used to scan and analyze the densitometry of the Western blot bands. Ten mice per group were used for behavioral data, of which five mice per group were used for either Western blot or immunofluorescence staining. All data are presented as the mean ± standard error of the mean. Statistical analyses and graphical representations were conducted using GraphPad Prism software (version 8.0, San Diego, CA, USA). Group comparisons were performed using one-way analysis of variance (ANOVA) followed by a Bonferroni post hoc multiple comparison test. A P value of less than 0.05 was considered statistically significant. Statistical significance is indicated as follows: *P < 0.05, **P < 0.01, and ***P < 0.001. ###P < 0.001. Symbols denote group comparisons: # indicates significance between control and Aβ1–42-treated group, while * denotes significance between the Aβ1–42 treated and Aβ1–42 + XA treated groups.
Comments (0)