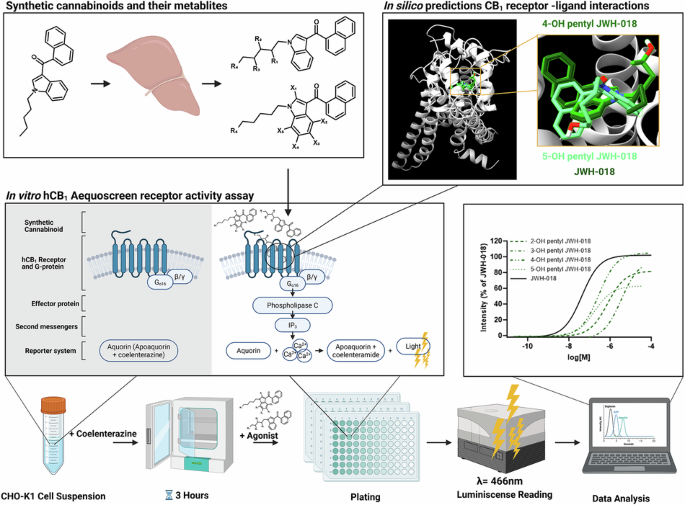
In this study, JWH-018, AM-2201, THJ-018 and THJ-2201 and their metabolites were screened for hCB1 efficacy and potency using a cell-based flash luminescence assay. This hCB1 receptor assay has previously been used to report on SC effects for both research purposes and to provide governmental agencies timely data aimed at speeding up the scheduling process of novel SC [8, 9, 18, 34].
In silico ligand-hCB1 simulations gave further indications on the behavior of the parent drug and metabolites within the receptor pocket. The molecular docking approach successfully reproduced the active conformation of the human CB1 receptor (hCB1) bound to the agonist methyl N-(1-((4-fluorobenzyl)-1H-indazole-3-carbonyl)-3-methyl-L-valinate (KCA), a co-crystallized ligand in the CB1 receptor structure (PDB ID: 6N4B), following remodeling to include the N-terminal domain [24]. The hCB1 receptor was remodeled in this study to ensure that likely interaction(s) between SC/metabolites and the N-terminal were not missed. RMSD value following simulation of the remodeled hCB1 and KCA residue was less than 0.9 Å demonstrating structural congruence.
It was found that the binding energy on the active form of the hCB1 receptor inversely correlates with the in vitro efficacy (Fig. 4). This finding indicates that the results from these two independent methods overlap, making it possible to extrapolate the in vitro results based on the in silico results. A weaker correlation between Ki and EC50 values was also observed (Supplementary Material B) which further strengthens this hypothesis. Thus, the predicted poses of the ligand in the hCB1 receptor after MD simulations could be used to further explain the differences in vitro efficacy and potency for each group of metabolites by looking at the number and type of interactions between the ligand and individual amino acids of the receptor. The found correlations between the in silico model and the experimental data suggest that this approach could be used for in depth studies of hCB1 activity of other SC and their metabolites too.
Structure-hCB1 activity relationship of JWH-018, AM-2201, THJ-018, THJ-2201
Comparing ligand configuration in the hCB1 receptor for the SC reveals only minor changes. All SC were full agonists with nanomolar-range potencies in vitro. Krishna et al., 2024, previously reported key interactions between JWH-018, and other derivatives from the JWH-018 family, and hCB1 using Schrödinger’s docking approach and molecular dynamics simulations. Similar results were obtained for JWH-018, THJ-018, AM-2201, and THJ-2201, depicting interactions with the amino acids, Phe170, Phe174, Phe200, and Trp279 are pivotal in SC stability in the hCB1 receptor pocket [35]. The largest difference observed in pairwise comparisons between AM-2201 and JWH-018 configurations was that AM-2201 had a more linear orientation in the hCB1 receptor pocket due to the fluorine atom at the 5th carbon of the pentyl chain. The addition of the electronegative fluorine on the tail interacts with His178, resulting in one additional amino acid interaction and perhaps anchoring AM-2201 in the pocket, contributing to the overall stability. This slight difference does not affect the configuration of the core and head, as the orientation of the substructures overlap. Similarly, the tail of THJ-2201 was more linear as compared to that of THJ-018. In general, the introduction of a fluorine atom stabilizes the molecule by enabling stronger binding to the receptor due to its high electronegativity and lipophilicity, thereby influencing the pharmacological effects of a drug [36].
Comparing the configuration of the two 5-fluoro-pentyl SC reveals that AM-2201 and THJ-2201 have overlapping orientation in the hCB1 pocket. The difference in their binding energy likely stems from the presence of two additional amino acid interactions for AM-2201 and better stabilization of the binding by the indole core of AM-2201 compared to the indazole core of THJ-2201 (Fig. 7). Additionally, Yano et al., 2023, demonstrated the interaction with His178 may not only stabilize 5-fluoro-pentyl SCRA derivatives in the hCB1 pocket, but also may be important in activating the CB1 receptor, in essence acting as an on/off switch for 5-fluoro-pentyl SC analogues [37]. All in all, chemical modifications more important than those above-mentioned need to be introduced for the SC to give a different effect in vitro.
Structure-activity comparison of hydroxypentyl metabolites
The length, bulkiness and constitution of the SC tail is found to be one of the key features to affect potency as well as efficacy [34]. 2-, 3- and 4-hydroxypentyl metabolites from all four SC in our study retained over 82.5% of the parent efficacy (Fig. 2). Moreover, we found that the addition of a hydroxyl group to the SC pentyl tail significantly reduces hCB1 potency in comparison to the parent. The exception seems to be the fluorinated 4-hydroxypentyl metabolites of AM-2201 and THJ-2201: for 4-hydroxypentyl AM-2201, the reduction in potency is minimal (1.7×), while for 4-hydroxypentyl THJ-2201 the potency increased (2.2×). Ranking the hydroxypentyl metabolites in order of potency reveals a different pattern (4-, 3-, 2-hydroxypentyl for AM-2201; 4-, 5-, 2-, 3-hydroxypentyl for JWH-018; 4-, 3-, 5-, 2-hydroxypentyl for THJ-018 and 4-, 3-, 2-hydroxypentyl for THJ-2201) compared to ranking them according to their efficacy or binding affinity.
Other in vitro studies previously reported JWH-018 as an agonist at nanomolar concentrations, and 4- and 5-hydroxypentyl JWH-018 as active metabolites, with 4-hydroxypentyl JWH-018 as the more efficacious and potent of the two. Pentanoic acid JWH-018 was found to be inactive in the same study [13]. These findings are consistent with the current study.
From our in silico results, there was not a clear amino acid interaction elucidating the underlaying mechanism of SC and their metabolites’ potency at the hCB1, although it is likely that multiplying interactions contributes to the stability of the compound within the receptor pocket, which in turn influences the compound’s potency based on the predicted Ki values.
A detailed analysis of the hCB1 predicted configurations for the hydroxypentyl metabolites reveals a trend attributable to the gradual shift (Fig. 7) of 2-, 3- and 4-hydroxypentyl metabolites in the hCB1 pocket. Interestingly, there was no noticeable difference between the R- and S- enantiomers of the metabolites. Regardless of this configuration, the metabolites in this study explicitly interacted with at least 8 amino acid in the hCB1 receptor pocket to be efficacious.
In contrast, both 5-hydroxypentyl metabolites caused a marked reduction in hCB1 efficacy and acted as a partial agonists, while the pentanoic acid metabolites were found to be inactive. In silico, the 5-hydroxypentyl and pentanoic acid metabolites of JWH-018 were oriented differently in comparison to JWH-018, as illustrated in Fig. 7, resulting in a reduced number of total interactions and a changed amino acid interaction pattern (Figs. 5, 6). Our results demonstrated that hydroxylation and carboxylation on the terminal carbon of the tail affect the efficacy, matching previously reported results from affinity [38] and efficacy experiments [13, 19, 39]. Notably, not all modifications on the fifth carbon of the tail seem to reduce efficacy. 5F-pentyl is one of the most common substructures for efficacious and potent SC and as discussed above, the pentyl and corresponding 5F-pentyl SC showed similar potencies in our study. The difference in observed efficacies could be due to the size, electronegativity and 3D structure of the different atoms or molecular groups. A hydroxyl group is larger than a fluorine atom despite having the same number of electrons. This is due to the high electronegativity of fluorine which keeps the electron cloud closer to the nucleus, also affecting the bond length (1.943 Å for C-OH bond and 1.400 Å for C-F bond) (see Supplementary Material B). Moreover, the F-C dipole is greater than the C-OH dipole. This depends both on the difference in electronegativity and the 3D geometry. The C-F bond is linear, while the C-OH bond shows a bent geometry. These differences lead to a marked change in ligand configuration comparing the 5-hydroxypentyl JWH-018 to AM-2201and could explain that the former act as partial agonist. All in all, the fluorine is both smaller and more polarized than the hydroxyl group. Addition of an alkene or a nitrile to the tail of the SC are other examples of modifications that retain both efficacy and potency and where the SC have been found to be involved in intoxications [40,41,42]. Both groups are planar, larger than fluorine but smaller in size and more electronegative than a hydroxyl group.
Structure-activity comparison of hydroxyindole metabolites
The hydroxyindole metabolites of JWH-018 and AM-2201 varied in potency, efficacy and binding affinities. The overall hydrophobic interactions between the ligand and the hCB1 receptor were reduced for the inactive metabolites such as the 7-hydroxyindole metabolites. Both in silico and in vitro data indicate that monohydroxyindole metabolites originating from AM-2201 are more potent than those from JWH-018 (1.9–3.8 times), although they are less potent than their parent. This finding is in line with previous literature on the differences in potency of pentyl/5-fluoropentyl SC [43].
Our results show that all 4-hydroxy and 5-hydroxyindole metabolites retained more than 88.9% of the parent hCB1 efficacy. The 6-hydroxy and 7-hydroxyindole metabolites retained efficacy for AM-2201, but showed reduced efficacy for JWH-018. To the best of our knowledge, in vitro data on hCB1 receptor activity is available for only two hydroxyindole metabolites of JWH-018. Cannaert et al. reported that 6-hydroxyindole JWH-018 had significantly less Emax than its 5-hydroxy isomer [13]. The result of this study is consistent with the previous data.
Both 2-hydroxyindole metabolites were inactive in vitro and showed matching low predicted binding energies and Ki, as well as fewer amino acid interactions in silico. The results of the SC metabolites tested in our study were consistent with the fact that all substructures of the molecule are involved in stabilizing the ligand in the hCB1 active site. Therefore, efficacy depended on the overall interactions within the hCB1 pocket rather than a single interaction. An example is seen when comparing the configurations of 7-hydroxyindole AM-2201 and 7-hydroxyindole JWH-018, where the 5-fluoro-pentyl tail stabilized the molecule in such a way that its terminal position after dynamics simulations was markedly different when compared to 7-hydroxyindole JWH-018 (Fig. 7).
Implications, effects, and toxicity
To the best of our knowledge, this study is the first investigating potency of a large set of metabolites from a SC with the same in vitro hCB1 activity method and in combination with docking and molecular dynamics simulations. In total, 81% of the metabolites were found to activate the hCB1 receptor. The 4-hydroxypentyl, 5-hydroxypentyl and pentanoic acid metabolites are reported as major metabolites of JWH-018, AM-2201, THJ-018 and THJ-2201 [10, 44, 45]. In this study, the 4-hydroxypentyl metabolites were among the most potent metabolites, whilst acting as full agonists, whereas the 5-hydroxypentyl metabolites were less potent and were partial agonists. This finding suggests that metabolites contribute to the pharmacodynamic effects of SC and possibly prolonging their effects.
As previously mentioned, 5-hydroxypentyl JWH-018 is a metabolite whose structure can be formed from both JWH-018 and AM-2201, the latter undergoing defluorination during metabolism [45]. Similarly, 5-hydroxypentyl THJ-018 and the pentanoic acid metabolite of THJ-018 can be formed from both THJ-018 and THJ-2201 [44]. According to Wolfarth et al. 2015, there is a distinct difference in the metabolic patterns of SC with a pentyl tail versus a 5-fluoro pentyl tail, with the ratio of formed 4-hydroxypentyl/5-hydroxypentyl metabolites differing. During metabolism, SC with a pentyl tail seem to be predominantly hydroxylated at the fourth position over the fifth carbon of the pentyl tail [46]. Together with our results, this might indicate a possible prolonged effect of the pentyl tailed SC over the 5F-analogs. However, this needs to be confirmed by further studies as 5-hydroxypentyl metabolites are readily oxidized to the pentanoic acid via an aldehyde intermediate [47]. This metabolic route seems to be one of the major detoxification pathways to eliminate both pentyl and 5-flouro pentyl SC, as the pentanoic acids are inactive at the hCB1 receptor. To fully understand the pharmacological effect of the SC, other factors such as frequency of use, amount of dosage and clearance among other factors should also be considered.
In summary, we show that several prevalent phase I metabolites of JWH-018, AM-2201, THJ-018 and THJ-2201 activated the hCB1 receptor in vitro as agonists with efficacies and potencies comparable to the respective SC. Structure and activity relationship of positional isomers show that metabolic pathways resulting in 5-hydroxypentyl metabolites and pentanoic acid metabolites lead to a decrease in hCB1 activity, with the former acting as partial agonist and the latter being inactive. The efficacy data from in silico experiments correlated with the in vitro results demonstrating a linear trend between the binding affinity and efficacy of the compounds investigated. This correlation as well as the ability to explain the experimental data based on shifting binding poses validates the in silico model as a useful tool to model hCB1 binding to SC and their metabolites. Our data show that the efficacy and potency of the SC and their metabolites seem to be driven by a complex network of hydrophobic weak amino acid-ligand interactions. This study highlights that oxidation to 5-hydroxypentyl and the inactive pentanoic acid metabolites is likely an important mechanism for SC detoxification. In contrast, 4-hydroxypentyl metabolites retain both efficacy and potency and likely contribute to overall SC effects upon intake and possibly the duration of these cannabinergic effects. Additionally, the present study not only expound our understanding of SCRAs and their metabolites’ activity at the molecular level, but also presents a rapid and comprehensive model to enable clinical and forensic toxicologists, and public health advocates to respond timely to the constantly evolving and dynamic SCRA landscape, and NPS in general.

Comments (0)